
胼胝体增生症
最近審查:12.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
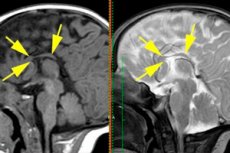
胼胝体发育不全是指连接大脑半球的神经纤维完全或几乎完全缺失的发育缺陷,它与胼胝体发育不全是同义词,即发育过程中缺乏形成。[ 1 ]
流行病學
脑部先天性畸形占胚胎发育期所有异常的至少25%。
据一些资料显示,在根据指征接受脑部MRI检查的患者中,有0.3-0.7%被检测出胼胝体异常,包括胼胝体发育不全(发育不全)。
儿童单独性胼胝体发育不全是一种罕见的先天性缺陷,但作为遗传性综合症的一部分,它被认为是一种相当常见的先天性异常,每 10,000 名发育障碍儿童中估计有 230 例。
在三分之一的胼胝体发育不全或部分发育不全病例中,会出现精神障碍。
原因 胼胝体发育不良
胼胝体发育不全(负责大脑两个半球之间的沟通和协调功能)是一种先天性缺陷,在大多数情况下,医生无法确定其确切病因。但最常见的是影响胎儿大脑结构宫内形成的染色体异常,或属于脑畸形遗传综合征的遗传异常。[ 2 ]
因此,在患有额外染色体综合征(三体综合征)的情况下,胎儿不会形成胼胝体,其中包括 Warkany 综合征、Patau 综合征和 Edwards 综合征。
胼胝体缺失见于遗传性莫瓦特-威尔逊综合征、艾卡迪综合征、马登-沃克综合征;唐-巴罗综合征、安德曼综合征、普劳德综合征、阿佩尔综合征以及X连锁脑积水综合征。胼胝体部分发育不全是皮特-霍普金斯综合征、丹迪-沃克综合征和森森布伦纳综合征的特征。
在脑回异常(如脑裂畸形)、先天性脑膨出和脑结构囊肿(如 Chudleigh-McCullough 综合征)以及畸形或Arnold-Chiari 综合征的情况下,胼胝体的形成会受到干扰。[ 3 ]
風險因素
胼胝体发育不全和其他先天性脑缺陷的可能风险因素包括:辐射增加和各种毒素对胚胎的致畸作用;怀孕期间饮酒和服用药物;使用某些药物和母亲的病毒感染。
如果家族有发育障碍和脑发育不良病史,孩子患上这种缺陷的风险也会增加。
發病
胼胝体在妊娠第六至第八周开始形成,但该过程的障碍可能在妊娠第三至第十五周之间发生。在胚胎学中,胼胝体缺失的发病机制与两种生物学机制相关。
首先,这可能由调控和协调背外侧迁移(胚胎细胞从神经嵴(神经管边缘的外胚层细胞带)或头部中内胚层向脑结构形成部位移动)的基因缺陷所致。大多数胚胎畸形和出生缺陷都是由于该过程中断造成的。
胼胝体发育不全的另一种机制可能是,新皮质神经元的轴突没有穿过胚胎大脑半球之间的中线,并且没有在左右大脑半球之间形成纤维束,而是形成了纵向分布的异常神经纤维束,而没有连接大脑半球。[ 4 ]
並發症和後果
胼胝体发育不全的后遗症和并发症取决于相关的脑部畸形。患有最严重脑畸形的儿童可能会出现癫痫、痉挛、脑积水以及身心发育障碍。
鑑別診斷
为了识别和鉴别胼胝体的其他病变,例如胼胝体发育不良(发育缺陷)、发育不全(部分发育不全)、胼胝体萎缩或发育不全,以及确认是否存在遗传综合症,需要进行鉴别诊断。[ 9 ]
誰聯繫?
治療 胼胝体发育不良
目前尚无方法将胼胝体恢复到正常状态。因此,治疗可能包括减轻该缺陷症状的严重程度:
- 使用抗惊厥药;
- 物理治疗,电休克疗法(增强肌肉力量,改善动作协调性);[ 10 ]
- 言语治疗;
- 通过职业治疗发展基本技能。
預防
作为预防措施,只能考虑预防各种因素的致畸作用和先天性疾病的产前诊断。
預測
总的来说,预后取决于胼胝体发育不全的表现程度和严重程度,以及是否存在伴随的发育缺陷。[ 11 ]
对于患有这种先天性异常的轻度儿童来说,其负面神经精神后果可能很小,他们的功能几乎正常。成年后,一些没有胼胝体的人智力正常,生活也正常。