
遺傳性腎炎(Alport綜合徵)在兒童中的應用
最近審查:23.04.2024

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
遺傳性腎炎(Alport綜合徵) - 遺傳決定的非免疫遺傳性腎小球病表現出血尿(有時蛋白尿),逐漸減少在腎功能的慢性腎功能衰竭發展通常與感覺神經性耳聾和視力受損相關聯。
LGGuthrie在1902年第一次描述了這種疾病,LGGuthrie觀察了幾代人中發現有血尿的家族。1915年,同一家AFHurst家族的成員描述了尿毒症的發展。1927年,阿爾波特首次發現了幾例伴有血尿的親屬的耳聾,在上世紀50年代,在這種疾病中描述了眼睛受傷。1972年,在遺傳性血尿患者中,形態學檢查腎組織,Hinglais等 顯示腎小球基底膜不均勻擴張和分層。1985年,遺傳性腎炎的遺傳基礎 - 一種IV型膠原基因突變(Fiengold等,1985)被確定。
通過對疾病遺傳性質的調查,可以得出結論:遺傳性腎炎(有或沒有聽力損失)的表型表現差異是由於突變基因的表達程度所致。因此,目前所有的臨床變體都被認為是一種疾病的表現,術語“遺傳性腎炎”與術語“Alport綜合徵”同義。
根據流行病學研究,遺傳性腎炎以每10萬名兒童17人的頻率發生。
Alport綜合徵的原因
該疾病的遺傳基礎是IV型膠原鏈的基因a-5中的突變。這種類型的通用的基礎用於腎臟膜,耳蝸設備,透鏡膠囊,視網膜和角膜,在使用單克隆抗體針對膠原級分的研究示出。最近,他們指出使用DNA探針進行遺傳性腎炎的產前診斷的可能性。
強調使用DNA探針檢測所有家族成員以鑑定突變基因的攜帶者的重要性,這對於對患有該病的家族進行醫學遺傳諮詢非常重要。然而,多達20%的家庭沒有親屬患有腎臟疾病,這表明異常基因中自發突變的發生率很高。大多數家族遺傳性腎炎患者患有腎臟疾病,聽力喪失和視力病理學; 因為相關個體的婚姻增加了從父母雙方獲得相同基因的可能性,所以有一個或多個祖先的人之間的相關婚姻。建立了與傳遞途徑的X染色體相關的常染色體顯性和常染色體隱性和顯性。
兒童更可能區分遺傳性腎炎的三種變體:Alport綜合徵,無聽力喪失的遺傳性腎炎和家族性良性血尿。
Alport綜合徵-聽力受損的遺傳性腎炎。基礎是腎臟腎小球基底膜膠原結構,耳和眼結構的組合缺陷。經典Alport綜合徵的基因位於X染色體長臂的21-22q位置。在大多數情況下,它是由與X染色體連鎖的顯性類型遺傳的。在這方面,在男性中,Alport綜合徵更加困難,因為在女性中,突變基因功能由第二完整染色體的健康等位基因補償。
遺傳性腎炎發展的遺傳基礎是IV型膠原α鏈基因的突變。這是被稱為IV型膠原蛋白的G六鏈:A5和A6基因鏈(Sol4A5和Sol4A5)位於在21-22q區X染色體的長臂上; 第2染色體上的a3-和a4-鏈(Co4A3和Co4A4)的基因; a1-和a2-鏈(Co4A1和Co4A2)的基因 - 在第13條染色體上。
在大多數情況下(80-85%),由於缺失,點突變或剪接紊亂,X連鎖型疾病遺傳與Co4A5基因的損傷相關。目前,發現了超過200個基因Kol4A5的突變,負責違反合成Ⅳ型膠原的α5鏈。在這種類型的遺傳中,疾病表現在兩性的孩子身上,但在男孩中則更為困難。
Co4A3和Co4A4基因位點的突變,負責合成Ⅳ型膠原的a3和a4鏈,是常染色體遺傳的。據研究顯示,16%的遺傳性腎炎,常染色體隱性遺傳的患者中有6%的患者有常染色體顯性遺傳。Co4A3和Co4A4基因有大約10個突變。
突變的結果違反了IV型膠原的裝配過程,導致其結構中斷。IV型膠原蛋白是腎小球基底膜,耳蝸器官和眼睛晶狀體的主要成分之一,其病理將在遺傳性腎炎的臨床中揭示。
IV型膠原,腎小球基底膜的組成部分,基本上由兩條鏈A1(IV)和一個A2鏈(IV),並且還包含A3,A4,A5鏈。最常當X連鎖遺傳Sol4A5突變伴隨缺乏A3,A4,和在結構IV型膠原的A6 A5鏈,並且O1和a2鏈的數目成腎小球基底膜增加。這種現象的機制尚不清楚,推測原因是轉錄後mRNA的變化。
缺乏A3,A4,和在該自身表現臨床最血尿(有時血尿或蛋白尿僅蛋白尿)Alport綜合徵的早期階段的薄型化和脆弱腎小球結果的結構IV型膠原基底膜A5鏈,聽力損失和圓錐形晶狀體。該疾病的進一步發展導致增厚,並且在該疾病的後期基底膜滲透性的破壞,在這些類型的膠原蛋白V和VI,表現在蛋白尿的增加和腎功能降低的生長。
遺傳性腎炎潛在的突變性質很大程度上決定了它的表型表現。當X染色體缺失的序列與同時突變和負責IV型膠原的A5和A6鏈的合成Sol4A6 Sol4A5基因,與Alport綜合徵食管平滑肌瘤病和生殖器組合。根據與缺失相關Sol4A5基因突變的研究標誌著大程度的病理過程,與腎損傷腎外表現和慢性腎功能衰竭的早期發展相結合,這種基因相比stochechnoy突變。
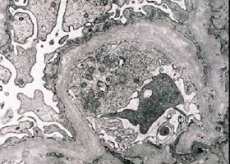
形態學上,電子顯微鏡顯示腎小球基底膜(尤其是椎板密度)的變薄和分層以及電子緻密顆粒的存在。腎小球損害在同一位患者中可能不均勻,從系膜最小局灶性病變至腎小球硬化。Alport綜合徵中的腎小球炎總是免疫陰性,與腎小球腎炎有區別。特徵是發育成管萎縮,淋巴組織細胞浸潤,存在含有脂質包裹物的“泡沫細胞” - 脂質體。隨著疾病的進展,基底腎小球膜的增厚和明顯的破壞被揭示。
揭示了免疫系統狀態的某些變化。患者遺傳性腎炎的Ig A的水平降低,並增加了IgM抗體的血液濃度的傾向,IgG的水平可以在疾病和下降在後期階段的早期階段增加。也許IgM和G濃度的增加是對IgA缺陷反應的一種補償反應。
T淋巴細胞系統的功能活性降低; 在負責IgA合成的B淋巴細胞中有選擇性減少,免疫的吞噬連接被破壞,主要是由於侵害嗜中性粒細胞的趨化性和細胞內消化
在腎活檢患者Alport綜合徵通過電子顯微鏡研究中,超微結構的變化觀察到的腎小球基底膜:變薄,分裂模式違反腎小球基底膜與在其厚度和不均勻的輪廓的變化。在遺傳性腎炎的早期階段,缺陷決定了腎小球基底膜的變薄和脆性。
腎小球膜變薄是更有利的跡象,在女孩中更常見。遺傳性腎炎中更常見的電子顯微鏡特徵是基膜的裂解,其破壞的嚴重程度與該過程的嚴重程度相關。

兒童Alport綜合徵的症狀
孤獨性尿綜合徵形式的Alport綜合徵的首發症狀在生命的前三年的兒童中更常見。在大多數情況下,這種疾病是偶然發現的。進入兒童醫療機構或ARVI期間,在對孩子進行預防性檢查期間,泌尿系統綜合徵將被揭示。如果在ARVI過程中出現尿液中的病理情況。在遺傳性腎炎中,與獲得性腎小球腎炎不同,沒有潛伏期。
在疾病的最初階段,孩子的幸福感很小,其特徵是尿綜合徵的持續性和持久性。其中一個主要症狀是不同程度的血尿,在100%的病例中觀察到。在呼吸道感染,體力消耗或預防性疫苗接種期間或之後注意到血尿的程度增加。蛋白尿在大多數情況下不超過1克/天,在疾病發作時可能不穩定,隨著過程進展蛋白尿增加。定期地,尿沉渣可能具有以淋巴細胞為主的白細胞尿,這與間質改變的發展有關。
後來,這違反了腎臟的部分功能,使患者的一般狀況惡化:中毒,肌肉無力,動脈低血壓,經常聽力受損(特別是男孩),有時視力受損。中毒表現為蒼白,疲勞,頭痛。在疾病的最初階段,大多數情況下的聽力損失僅通過聽力檢查來檢測。聽力損失可能發生在兒童不同時期,但大多數聽力損失在6-10歲時被診斷出。兒童的聽力損失始於高頻率,在空氣和骨骼傳導方面達到相當程度,從聲音傳導到聽覺聾。聽力損失可能是該疾病的首要症狀之一,可能會導致泌尿系統綜合症。
在20%的病例中,Alport綜合徵患者的眼睛有變化。鏡片最常見的異常:spherofokiya,lentikonus前,後或混合,各種白內障。在Alport綜合徵家庭中,近視發生率很高。許多研究人員經常在這些家庭中註意到黃色身體區域明亮的白色或淡黃色顆粒形式的雙側週期性變化。他們認為這種症狀是一種常見症狀,對Alport綜合徵具有較高的診斷價值。C. S. Chugh et al。(1993),用於眼科研究揭示Alport綜合徵患者在箱子66.7%視力下降,前向圓錐形晶狀體 - 37.8%,視網膜上的點 - 在22.2%,白內障 - 20%,圓錐角膜 - 6 ,7%。
在一些遺傳性腎炎患兒,尤其是腎功能不全患者中,發現身體發育明顯滯後。隨著腎功能不全的發展發展成高血壓。在兒童中,青春期和年齡較大的人群更容易發現。
特徵是患有遺傳性腎炎的各種(超過5-7)柱狀結締組織染色體發育的患者存在。在柱頭患者結締組織最常見的眼增寬,高腭,牙頜畸形,異常形狀的耳朵,他的手,“sandalevidnaya差距”對腳小指的曲率。對於遺傳性腎炎是家庭,以及其分佈的中先證者的親屬的頻率高,通過該疾病被傳輸內的特徵在於均一性dizembriogeneza柱頭。
在疾病的早期階段發現的分離的部分減少的腎功能:的氨基酸,電解質,濃度功能酸化運輸,進一步的變化是在近端和遠端腎單位兩者的功能狀態和具有結合部分的病症的性質。腎小球濾過的減少發生在晚些時候,更常見於青春期。隨著遺傳性腎炎的發展,貧血發展。
因此,對於疾病的遺傳性腎炎特徵分期:第一潛階段或由最小的變化性膀胱綜合徵表現隱藏臨床症狀則發生逐漸失代償過程與腎功能的降低有明顯的臨床症狀(中毒,無力,發育遲緩,anemizatsiya)。臨床症狀通常與炎性反應的分層無關。
遺傳性腎炎可以表現在不同的年齡段,這取決於基因的作用,直到特定的時間處於壓抑狀態。
分類
有三種遺傳性腎炎的變種
- 我變種 - 臨床上表現為腎炎伴血尿,聽力下降和眼睛損傷。隨著慢性腎功能衰竭的發展,腎炎的病程進展。遺傳類型是顯性的,與X染色體有關。形態上,基底膜的結構,它的變薄和劈裂是紊亂的。
- II型 - 在臨床上表現為腎炎伴血尿,無聽力下降。隨著慢性腎功能衰竭的發展,腎炎的進展是漸進的。遺傳類型是顯性的,與X染色體有關。形態學上,顯示腎小球毛細血管(特別是層狀)的基膜變薄。
- III選項 - 良性家庭血尿。療程有利,慢性腎功能衰竭不發展。遺傳類型是常染色體顯性遺傳或常染色體隱性遺傳。在常染色體隱性遺傳類型中,女性患有更嚴重的疾病。
Alport綜合徵的診斷
建議採用以下標準:
- 在至少兩名腎病患者的每個家庭中存在;
- 血尿是先證者腎病的主要症狀;
- 至少有一名家庭成員有聽力損失;
- 慢性腎功能衰竭發生在一個親戚和更多。
在各種遺傳和先天疾病診斷的重要場所屬於一個綜合的方法來檢查,首先在關注孩子的血統的製備中獲得的數據。診斷綜合徵奧爾波特視為有效在患者3出來的4個典型特徵的情況:在電子顯微鏡表徵活檢標誌檢測在家庭血尿和慢性腎功能衰竭的存在,患者的感覺神經性聽力損失的情況下,病狀裂解腎小球基底膜與它的厚度的變化和不均勻的輪廓。
對患者的檢查應包括臨床遺傳學調查方法; 定向研究疾病的病史; 考慮診斷標準的患者的一般檢查。補償階段病理能趕上只關注這種綜合徵具有家族病史,低血壓,多柱頭dizembriogeneza改變膀胱綜合徵。在失代償estrarenalnyh可能引起的症狀,如嚴重中毒,無力,遲鈍的身體發育anemizatsiya表現和腎功能的逐漸減少放大。在大多數腎功能下降的患者中,觀察到酸和氨基酸功能的降低; 50%的患者註意到腎臟分泌功能顯著下降; 限制尿液光密度波動的範圍; 違反過濾節律,然後腎小球濾過率下降。慢性腎功能衰竭的階段在患者中診斷為3-6個月並且血清尿素水平升高(超過0.35g / l),腎小球濾過率降低至標準的25%。
遺傳性腎炎的鑑別診斷必須與所獲取的表單hematuric腎小球腎炎主要進行。已經獲得了開始的先前感染後2-3週期間日益急性腎小球腎炎,腎外的功能,包括與所述第一天高血壓(在遺傳性腎炎,相反地,低血壓),在發病腎小球濾過率降低,沒有違反的部分管狀的功能,而就像它們存在的遺傳一樣。收購腎小球腎炎與更嚴重的血尿,蛋白尿,與血沉增快發生。腎小球基底膜特徵性遺傳性腎炎的典型變化具有診斷意義。
代謝障礙性腎病的鑑別診斷與鑑定臨床蒙納腎臟病家族慢性腎功能衰竭進行,範圍可以從腎病腎盂腎炎到尿路結石。兒童經常抱怨腹痛,並定期尿尿,在尿沉渣 - 草酸鹽。
如果您懷疑遺傳性腎炎患者應該去專科腎臟科就診。
需要檢查什麼?
如何檢查?
需要什麼測試?
誰聯繫?
治療Alport綜合徵
在政權規定限制大量體力消耗時,留在新鮮空氣中。飲食高檔,含有足量的高檔蛋白質,脂肪和碳水化合物,兼顧腎臟的功能。慢性病灶感染的鑑定和康復非常重要。從藥物,ATP,輔酶,吡哆醇(高達50毫克/天),使用肉鹼氯化物。課程每年舉辦2-3次。當血尿是規定植物療法 - 蕁麻,蕁麻,黑莓灰,蓍草。
在國外和國內文獻中報導了用潑尼松龍治療和使用細胞抑製劑。但是,效果很難判斷。
在慢性腎功能衰竭中,使用血液透析和腎移植。
沒有特定的(有效的發病)治療遺傳性腎炎的方法。所有醫療措施都旨在預防和減緩腎功能的減退。
考慮到腎臟的功能狀態,飲食應當平衡並且高熱量。在沒有侵犯兒童營養功能狀態的情況下,應該有足夠的蛋白質,脂肪和碳水化合物含量。在存在腎功能障礙徵兆的情況下,蛋白質,鈣和磷的碳水化合物含量應受到限制,這會延緩慢性腎功能衰竭的發展。
身體上的壓力應該是有限的,建議孩子不要做運動。
避免接觸傳染病患者,降低發生急性呼吸道感染的風險。有必要消毒慢性感染灶。沒有進行遺傳性腎炎兒童的預防性接種疫苗,只能根據流行病學指徵接種疫苗。
荷爾蒙和免疫抑制治療遺傳性腎炎是無效的。多年來長期使用環孢素A和ACE抑製劑有一定的積極作用(減少蛋白尿並減緩疾病進展)。
在使用改善代謝的藥物治療患者中:
- 吡哆醇-2-3mg / kg /天,3次分劑量,持續4週;
- kokarboksilaza - 每隔一天肌肉注射50毫克,僅注射10-15次;
- ATP - 每隔一天肌內註射1ml,注射10-15次;
- 維生素A - 1000 U /年/天1次接待2週;
- 維生素E - 1 mg / kg /天1次接待2週。
這種治療改善了患者的一般狀況,減少了腎小管功能障礙,並且每年給藥3次。
由於免疫調節劑可能使用左旋咪唑-2毫克/千克/天,每週2-3次,間隔3-4天劑量間隔。
對研究人員來說,高壓氧對血尿和腎功能不全的嚴重程度有積極作用。
治療遺傳性腎炎最有效的方法是及時腎移植。在移植中沒有發生疾病復發,在少數病例中(約5%),移植腎中與抗原相關的腎炎在腎小球基底膜上發展是可能的。
一個有前途的領域是產前診斷和基因工程治療。對動物的實驗顯示將負責合成IV型膠原α鏈的正常基因轉移至腎組織的高效率,然後注意到正常膠原結構的合成。
Использованная литература