
儿童和成人安杰曼综合征
最近審查:04.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。

有些疾病,用“好好照顾自己就不会生病”这样的话来形容,至少听起来很荒谬。这些疾病是指孩子在出生前就存在一些精神和身体上的先天异常,但这并非父母的错。这类疾病是由染色体组的突变或异常引起的,被称为染色体疾病或遗传疾病。安杰尔曼综合征、唐氏综合征、帕陶综合征、爱德华兹综合征、特纳综合征、普拉德-威利综合征——这只是相当多遗传疾病中的一部分。
快乐男人综合症
这次我们将讨论一种以英国儿科医生哈里·安吉尔曼(Harry Angelman)命名的病理学。他于1965年首次提出了这个问题。此前一天,他在诊疗过程中遇到了三个不同寻常的儿童,他们有着共同的奇特症状。这位医生将这些孩子称为“玩偶儿童”,并撰写了一篇关于他们的文章,最初名为《儿童木偶》。文章本身及其标题灵感源自维罗纳一家博物馆中展出的一幅画作。这幅画描绘了一个正在大笑的男孩,名为《木偶男孩》。画中的孩子与安吉尔曼在诊疗过程中遇到的三个孩子之间存在关联,这促使这位儿科医生根据他们所患的疾病将这三个孩子归为一类。
文章中提到的孩子没有引起其他医生的注意,这并不奇怪。毕竟,乍一看,他们似乎患有完全不同的疾病,而且这三个不同病例的总体临床表现差异巨大。或许这种“新”染色体病理学会引起其他科学家的兴趣,但当时遗传学技术还不够发达,无法证实这位英国医生的假设。因此,在引起人们一定兴趣之后,这篇文章被搁置了很长时间。
第二次提及“安吉尔曼综合征”(Angelman syndrome),也就是英国儿科医生G. Angelman发表的那篇文章,可以追溯到20世纪80年代初。直到1987年,人们才找到原因,为什么一小部分孩子天生就有这种畸形,以至于从外表看,他们似乎总是面带微笑,快乐无比。事实上,这根本不是事实,他们的笑容只是一种苦笑,背后隐藏着不快乐的人类灵魂和父母的痛苦。
原因 安杰曼综合征
天使综合征(Angelman syndrome)是一种染色体病变的医学名称,但它远非唯一的一种。人们把这种疾病称为“娃娃儿童综合征”、“快乐木偶综合征”、“彼得鲁什卡综合征”和“笑娃娃综合征”。人们想出了各种各样的名字(有时甚至冒犯了患者本人及其父母),但疾病就是疾病,无论它看起来多么滑稽,也无论其原因是什么。
天使综合征的病因,与许多其他遗传性疾病一样,在所有情况下,都是由一条染色体或整个染色体组的结构紊乱引起的。但就我们的情况而言,问题出在从母亲遗传下来的15号染色体上。也就是说,在本例中,父系染色体没有出现异常,但母系染色体发生了某些突变。
根据染色体异常的类型,Angelman综合征被归类为染色体突变。此类突变被认为是:
- 缺失(缺少包含某组基因的染色体部分;如果其中一个基因缺失,则称为微缺失),这是原始染色体部分丢失时两次断裂和一次重新连接的结果。
- 重复(染色体中存在一个额外的部分,该部分是现有染色体的副本),在大多数情况下会导致人死亡,较少情况下会导致不孕。
- 倒位(将染色体的某一部分反转 180 度,即朝相反的方向,此时其中的基因位于相反的顺序),是指染色体断裂的末端以与原来不同的顺序连接起来。
- 插入(如果染色体中的部分遗传物质错位),
- 易位(如果染色体的某个部分附着在另一条染色体上;这种突变可以是相互的,而不会丢失任何部分)。
如果孩子从毫无戒心的母亲那里获得突变的染色体,那么孩子注定会出生时就存在畸形。目前,天使综合征最常见的病因仍被认为是母体第15条染色体的缺失,也就是一小部分缺失。“笑娃娃”综合征中不太常见的突变被认为是:
- 易位,
- 单父二体(如果孩子从父亲那里得到一对染色体,则缺少母体染色体),
- DNA 中基因的突变,DNA 既是主要的构建(遗传)材料,也是其正确使用的指令(特别是母体染色体中 ube3a 基因的突变)。
父母存在上述突变之一,是儿童患天使综合征的风险因素。但不仅是染色体突变,基因组突变(与染色体组的数量变化相关,比染色体突变更常见)也可能引发儿童患上该病。常见的基因组突变包括染色体三体性(即一个人的染色体组超过46条染色体)。
孩子出现某种病症,并不一定需要父母有染色体异常。然而,仍有一定比例的患者患有遗传性疾病。
發病
让我们更深入地探讨一下生物学,或者更准确地说,遗传学。每个人体的遗传信息都包含在23对染色体中。一对染色体中的一条由父亲遗传给孩子,另一条由母亲遗传给孩子。所有染色体对的形状和大小各不相同,并携带着特定的信息。因此,第23对染色体(X和Y染色体)负责婴儿性别特征的形成(XX代表女孩,XY代表男孩,而Y染色体只能由孩子从父亲那里获得)。
理想情况下,孩子会从父母那里获得46条染色体,这些染色体构成了他的遗传特征,预先决定了他作为个体的特质。染色体数量过多被称为三体性,被认为是偏离常态的表现。例如,染色体组(核型,决定物种和个体特征)中存在47号染色体会导致唐氏综合征的发生。
如果染色体用特殊染料染色,在显微镜下可以看到每条染色体上都有不同深浅的条纹。每条条纹内都包含大量基因。所有这些条纹都由科学家编号,并有固定的位置。缺失其中一条条纹就被认为是异常。在安格曼综合征中,人们经常可以观察到位于长臂q11-q13区间的母体染色体片段缺失,该区间的DNA碱基数量仅为约400万个。
染色体的主要成分被认为是一个极其长的DNA分子,包含数千个基因和数亿个含氮碱基。因此,负责天使综合征和其他几种疾病发展的15号染色体包含1200个基因和约1亿个碱基。DNA分子结构的任何紊乱都必然会影响未来孩子的外貌和发育。
基因中包含的遗传信息被转化为蛋白质或RNA。这个过程被称为基因表达。通过这种方式,从父母处获得的遗传信息既具有形式,也具有内容,并体现在其独特的女性或男性继承人身上。
有许多病理具有非经典的遗传类型,包括天使综合征,其中从父母那里获得的成对染色体的基因带有父母的独特印记,并以不同的方式表现出来。
因此,天使综合征是基因组印记的一个显著例子,其基因在儿童体内的表达直接取决于等位基因来自哪一方(同一个基因的不同形式,分别来自父亲和母亲,位于成对染色体的相同区域)。也就是说,只有母亲染色体的异常才会导致该综合征,而父亲染色体的突变和结构异常则会导致完全不同的病理。
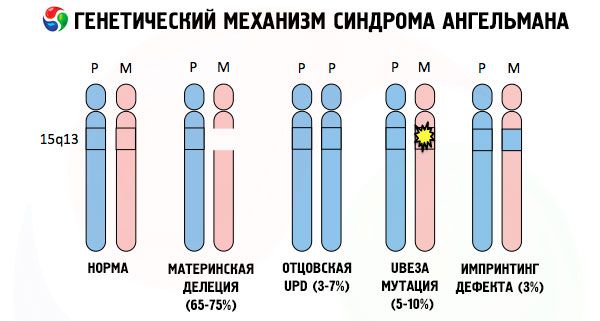
这种病症是指母体染色体中某些基因缺失,或单个基因活性丧失/降低(在绝大多数情况下,是ube3a基因,该基因参与泛素的代谢,泛素是一种调节其他蛋白质降解的蛋白质)。因此,儿童会被诊断出智力发育异常和身体畸形。
症狀 安杰曼综合征
天使综合征的症状会影响儿童生活和发育的各个方面:身体、神经和精神。基于此,可以识别出三组症状来指示该病症的发展。
- 外部或身体症状:
- 与正常大小的身体和四肢相比,头部过小,
- 嘴巴太宽,
- 脸上几乎总是挂着微笑(嘴巴张开),
- 牙齿稀疏,
- 上唇狭窄,
- 经常伸出宽大的舌头,
- 突出的下颌,
- 尖下巴,
- 皮肤非常白,通常有毛发(白化病,与身体不产生黑色素有关),
- 白皙皮肤上的黑斑(由于黑色素生成不足导致的色素减退)
- 身体或外部症状:斜视或视神经萎缩等眼部疾病,
- 脊柱弯曲(脊柱侧弯),
- 腿部僵硬(由于关节活动性低,走路时膝盖不能弯曲,因此与娃娃的步态相似)。
- 与心理和情感发展相关的症状:
- 严重智力障碍,
- 情绪过于激动、吵闹、挑剔的行为,
- 经常拍手,
- 表达友好,脸上始终挂着微笑,
- 经常无缘无故地大笑。
- 神经系统症状:
- 四肢震颤,
- 动作协调性不足,失去平衡,
- 肌肉张力下降,
- 各种睡眠障碍,
- 童年时期经常歇斯底里,
- 言语障碍(孩子开始说话较晚,沟通能力差,言语不清),
- 在兴奋性增加的背景下的多动症,
- 注意力不集中和学习困难。
但这仅仅是对该疾病的概括。事实上,安格曼综合征的临床表现很大程度上取决于疾病的发展阶段以及导致病理的染色体突变类型。这意味着该疾病的症状在不同患者中可能存在显著差异,长期以来,我们无法将其病理与其他具有相似临床表现的患者区分开来。
在所有症状中,我们可以突出那些无一例外地代表所有患者的症状:
- 严重智力障碍,
- 不当行为(无理笑声、兴奋增加、注意力不集中、欣快状态),
- 运动技能发育不良,
- 动作协调性差、步态共济失调(步伐不均匀、左右摇摆等)、四肢震颤。
- 言语发育障碍,以非言语交流方式为主。
在绝大多数患者遇到的症状中,可以区分出以下几点:
- 因身体发育迟缓而导致的头部与身体不成比例,
- 在许多患者中,头骨的形状使得大脑的尺寸比健康人的大脑尺寸要小(小头畸形),
- 3岁前癫痫发作,随着年龄增长,发作强度和频率逐渐下降,
- 脑电图参数失真(低频波的波动和高振幅)。
这些症状相当常见,然而,20% 的 Angelman 综合征患者并没有出现这些症状。
更不常见的是,可以诊断出该疾病的如下表现:
- 严重或轻度斜视,
- 舌头运动控制不佳,导致患者经常无缘无故伸出舌头,
- 吞咽和吸吮困难,尤其是在幼儿中,
- 破坏皮肤和眼睛的色素沉着,
- 走路时举起或弯曲手臂,
- 反射亢进,
- 睡眠障碍,尤其是在儿童时期,
- 经常流涎,
- 无法满足的口渴,
- 咀嚼动作过于活跃,
- 对热过敏,
- 后脑勺扁平,
- 突出的下颌,
- 光滑的手掌。
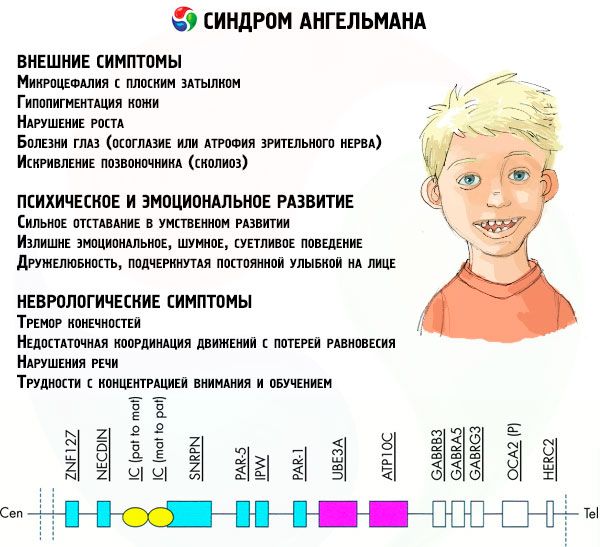
相当一部分患者存在排尿问题(排尿控制不佳)、精细运动技能受损(导致自理和学习困难)以及体重过重。几乎所有患者青春期发育都比健康同龄人晚。
患有天使综合征的儿童能够很好地感知并理解口语,但他们不愿参与对话,只能说出日常生活中必要的几十个单词。然而,成年后,这类患者看起来比没有遗传病的同龄人更年轻。
安格曼综合征的许多症状并不稳定,因此该病的临床表现会随着年龄的增长而发生显著变化。抽搐和癫痫发作的频率会降低甚至完全消失,患者变得不那么容易兴奋,睡眠质量也会提高。
並發症和後果
安吉尔曼综合征是一种严重的、目前几乎无法治愈的染色体病变,它剥夺了患者过上正常生活的机会。安吉尔曼综合征患儿的生活状况很大程度上取决于染色体异常的类型。
在大多数情况下,染色体片段的重复会导致生命不平衡。即使这类患者在婴儿期没有死亡,并且能够活到青春期,他们也没有生育孩子的机会。
天使综合征中最常见的部分基因缺失或缺失,会阻碍儿童学习走路和说话。这类儿童的智力障碍更为严重,癫痫发作的频率更高,且强度远高于其他染色体异常患者。
如果只有一个基因发生突变,只要给予适当的关注和方法,孩子就可以学会自我护理、沟通和群体互动的基本知识,尽管他在发展上仍然会落后于同龄人。
对于天生善良的安吉曼综合征患儿来说,父母的爱与关注至关重要。只有这样,孩子的教育才能结出硕果,即使成果有限。当然,安吉曼综合征患者无法在普通学校学习。他们需要参加特殊课程,首先学习如何集中注意力,然后逐步学习学校的基础知识。
診斷 安杰曼综合征
天使综合征是一种先天性发育障碍。但由于某些原因,在婴儿期和幼儿期通常无法诊断。这是因为该病在3岁以下婴儿和儿童中症状不具特异性且表现不明显。而且,该病在我国的发病率并不高,医生们尚未学会在同龄人中识别它。
婴儿期的安格曼综合征可表现为肌张力下降,进而导致喂养困难(吸吮和吞咽反射减弱),以及日后学步困难(这类儿童学步较晚)。这些症状是婴儿发育异常的早期迹象,很可能与染色体异常有关。只有基因分析才能证实这一假设。
父母患有各种基因或染色体异常的儿童尤其需要关注。毕竟,疾病最初可能不会显现,如果能及时发现病症,并开始与孩子密切合作,就有可能显著提高学习成绩,减缓病情的进展。
如果父母有各种染色体异常,甚至在婴儿出生前就会进行基因分析,因为 SA 是可以在胚胎阶段检测到的病理之一。
遗传研究材料的收集可以通过两种方式进行:
- 侵入性(具有一定的风险,因为需要穿透子宫才能采集羊水样本),
- 非侵入性(从母亲的血液中分析婴儿的 DNA)。
然后进行以下研究:
- 荧光原位杂交(FISH 方法)——将用特殊染料标记的 DNA 探针与所研究的 DNA 结合,然后在显微镜下检查。
- ube3a 基因和印迹基因突变分析,
- 使用遗传学中的特殊方法进行 DNA 甲基化分析。
基因检测在染色体异常的情况下能够提供相当准确的信息,这意味着未来的父母可以提前知道该如何应对。然而,也存在例外。在某些患者群体中,即使存在所有提示病理的症状,检测结果仍然正常。也就是说,只有通过从幼儿时期仔细观察孩子才能发现病理:比如他的饮食习惯、何时开始走路和说话、走路时是否弯曲双腿等等。
除了 FISH 方法外,在 Angelman 综合征的仪器诊断方法中,还可以选择断层扫描(CT 或 MRI)来帮助确定大脑的状况和大小,以及脑电图(EEG),它可以显示大脑各个部分如何工作。
医生通常在3至7岁时做出最终诊断,此时患者已出现大部分症状,且疾病发展的动态已清晰可见。
需要什麼測試?
鑑別診斷
天使综合征是一种几乎没有特异性表现的遗传性疾病。大多数症状既可以提示强直性脊柱炎(AS),也可以提示其他遗传性疾病。
天使综合征的鉴别诊断是通过以下病理进行的:
- 皮特-霍普金斯综合征(患者特征为智力低下,性格开朗,爱笑,嘴巴较大且宽,有小头畸形)。区别在于患者清醒时会出现过度换气和屏住呼吸的症状。
- 克里斯蒂安森综合征(患者为智力障碍者,性格开朗,无法说话,特征为小头畸形、共济失调、抽搐、不自主的肌肉运动)。
- 莫瓦特-威尔逊综合征(症状:智力低下、癫痫发作、尖下巴、嘴巴张开、面部表情喜怒无常、小头畸形)。特征:两眼间距大、眼球内斜、鼻尖圆、耳廓后翻。
- 歌舞伎综合征(特征包括轻度至中度智力障碍、言语和运动障碍、肌肉无力、癫痫发作、小头畸形、瘙痒间隔时间长以及协调性受损)。其特征为:眉毛拱起,下眼睑外侧部分外翻,眼距宽,睑裂长,睫毛浓密长。
- 雷特综合征(与女性强直性脊柱炎的区别)。症状:言语发育迟缓、癫痫发作、小头畸形。区别在于,患者面部没有快乐的表情,并伴有呼吸暂停和失用症发作,且病情会随着时间的推移而加重。
- 常染色体隐性智力障碍综合征38(症状:明显的智力障碍,运动技能和言语发育迟缓,肌肉无力,婴儿喂养问题,冲动)。其显著特征是虹膜呈蓝色。
- MECP 2基因重复综合征(与男性SA不同)。症状:严重智力障碍,自儿童期出现肌无力,言语障碍或失语,癫痫。特征:进行性肌病,持续反复感染。
- 克莱夫斯特拉综合征(症状:言语和思维障碍、肌肉无力、睡眠障碍、注意力不集中、张口、多动、癫痫、共济失调、平衡障碍)。其显著特征:扁平脸、短鼻、眼距宽、下唇外翻、易怒。
- 史密斯-马吉尼斯综合征(以癫痫、睡眠问题、智力和运动发育障碍为特征)。其显著特征包括宽而扁平的面部和突出的前额。
- 库伦-德弗里斯综合征(轻度至中度智力障碍、肌肉无力、癫痫发作、友善)。特征:长脸、高额头、招风耳、斜眼、关节活动度高、先天性心脏缺陷。
- 费兰-麦克德米德综合征(症状:智力低下、言语障碍或失语)。特征:手掌较大,肌肉发达,先天性肌肉无力,出汗能力弱。
腺苷酸琥珀酸缺乏症、常染色体隐性遗传性智力低下综合征 1、染色体 2q23.1 重复综合征、FOXG1、STXBP1 或 MEF2C 基因单倍体不足综合征等病症可以“夸耀”与天使综合征相似的症状。
医生的任务是做出准确的诊断,将天使综合征与具有类似症状的病理区分开来,并根据疾病的诊断阶段开出有效的治疗方法。
治療 安杰曼综合征
天使综合征是医学界仍在寻求有效治疗方法的病症之一。该病的病因治疗正处于各种方法和手段的开发阶段,其中许多尚未在人体上进行过测试。这意味着目前医生只能局限于对症治疗,这在某种程度上有助于缓解患有木偶综合征的儿童和成人的痛苦处境,他们饱受癫痫发作、唾液分泌、低血压和睡眠障碍的困扰。
因此,通过选择合适的抗惊厥药物,可以降低癫痫发作的频率和强度。但问题在于,SA患者的癫痫发作与普通癫痫发作不同,其特点是发作类型多样,这意味着可以通过同时使用多种药物来缓解病情。
用于治疗天使综合征的最常用抗惊厥药物包括:丙戊酸、托吡酯、拉莫三嗪、左乙拉西坦、氯硝西泮以及以这些药物为基础的药物。较少使用的药物包括以卡马西平、苯妥英钠、苯巴比妥和乙琥胺为基础的药物,因为其中一些药物可能会引发一种反常效应,即增强癫痫发作并增加其发作频率。如果将这些药物作为单药治疗的一部分,就会出现这种情况。
治疗流口水通常有两种方法:药物治疗(抑制唾液分泌的药物)和手术治疗(重建唾液管)。但对于唾液腺功能亢进症(SA),这些方法被认为是无效的,而且这个问题仍然悬而未决。家长和照顾此类患者的人员必须特别注意这个问题,因为患者本身通常无法控制流口水,有些甚至无法自理。
另一个问题是睡眠时间短。患有天使综合征的儿童通常睡眠时间不超过5小时,这会对全身机能产生负面影响。这些孩子容易兴奋、好动,喜欢游戏和交流(即使他们尽量限制自己使用非语言方式),但白天会明显感到疲倦。为了获得良好的休息,身体需要深度、充分的睡眠,但这恰恰是关键所在。
镇静药物(吩噻嗪类和非典型抗精神病药物)似乎足以改善易兴奋患者的睡眠。但对于强直性脊柱炎 (AS) 患者来说,使用此类药物很容易产生副作用。因此,医生仍然更倾向于使用温和的安眠药,例如褪黑素(一种基于睡眠激素的天然激素药物),患者睡前一小时服用一片,以及苯海拉明。医生会根据患者的病情和年龄决定给药频率和剂量。
天使综合征患者有时会出现消化和排便问题。您可以使用泻药(最好是草药)来改善排便。
或者,你也可以像美国医生那样,基于一些自闭症治疗方法,换一种思路来解决这个问题,因为许多强直性脊柱炎的典型症状也是自闭症的特征(冲动、不自主运动、重复性动作、注意力缺陷、沟通障碍等等)。有人指出,使用促胰液素这种激素,可以促进消化和排便的正常化,对患者的注意力有积极的影响,而催产素则有助于提高孩子的认知能力、记忆力和矫正行为。
诚然,单靠激素治疗是不够的,尤其对儿童而言。对于安琪儿综合征,需要行为疗法、与心理学家和言语治疗师合作(教授非言语交流方法和手语)。这类儿童的教育应以个性化计划为基础,由受过专门培训的教师、心理学家和家长共同参与。遗憾的是,并非所有地方都能做到这一点,家庭只能独自面对问题。
由于许多年轻的强直性脊柱炎患者存在肌张力低下和关节问题,物理治疗备受关注。医生通常会采用石蜡敷贴、电泳和磁疗等疗法。
积极的滋补按摩和特殊的治疗性体能训练有助于患儿在一段时间后能够站立并自信地行走。水中体操在这方面尤其有效,建议在冷水中进行。它可以增强肌肉张力,并教会孩子控制身体和协调动作。
抗惊厥治疗
安格曼综合征最危险的症状是类似癫痫的癫痫发作。80%的患者会出现这种症状,这意味着所有患者都需要接受有效的抗惊厥药物治疗。
癫痫发作的治疗需要维生素和抗惊厥药物。对于伴有惊厥综合征的安吉曼综合征,B族维生素以及维生素C、D和E可能会有所帮助。但在这种情况下自行开具维生素治疗非常危险,因为不加控制地服用维生素会降低抗癫痫药物的疗效,并引发新的、更严重、持续时间更长的癫痫发作。
抗惊厥药物的选择及其有效剂量也应由专科医生决定。专科医生还会决定一种药物是否足够,还是患者需要长期服用两种或两种以上的药物。
对于大多数患者,医生会开丙戊酸类药物(Valproic acid、Depakine、Convulex、Valparin等),以防止癫痫发作并改善患者的情绪和精神状态。
丙戊酸有片剂、糖浆和注射液等剂型。最常用的药物是缓释片“德巴金”,其剂型包括片剂和静脉注射液。医生会根据患者的体重、年龄和病情,具体确定药物剂量。
该药每日2至3次,随餐服用。平均每日剂量为每公斤患者体重20-30毫克,最大剂量为每天每公斤50毫克。
禁忌症。肝脏和胰腺功能障碍、出血素质、肝炎、卟啉症以及对本品过敏者禁用。
副作用包括手震颤、消化和排便障碍以及体重变化。
“托吡酯”也是治疗SA的首选药物。该药为片剂,既可作为单药治疗,也可与其他药物联合使用。
服用方法和剂量。口服片剂,与进食无关。成人初始每日剂量为25-50毫克,儿童初始每日剂量为0.5-1毫克/公斤体重。每周根据医生指示增加剂量。
孕妇、哺乳期妇女及对本品成分过敏者禁用。本品有多种副作用。
医生可能会为天使综合征开出的药物:氯马西泮、利沃曲尔、拉莫三嗪、Seizar、拉莫三嗪、左乙拉西坦、开浦兰、Epiterra 等。
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
传统医学和顺势疗法
传统药物,如顺势疗法药物,当然相对安全,但这种治疗对天使综合征的有效性仍存在争议。
虽然民间疗法在某些方面仍然有效。比如说,比如阻止癫痫发作。在这方面,草药治疗可能非常有效。
以牡丹、甘草和浮萍(各成分等量服用)为基础的药方效果良好。这些草药需要磨成粉。服用两周后,癫痫发作频率显著减少。
薰衣草煎剂(每杯开水加1茶匙)也有助于缓解痛经。将混合物煮沸5分钟,然后浸泡半小时。该药在晚上服用,持续14天。
益母草的水浸液(或酒精浸液)被认为对治疗癫痫发作有效。
在预防安吉曼综合征癫痫发作的顺势疗法药物中,可以使用以洋甘菊和益母草为基础的药物、氰酸、硝酸银、溴钾和白砷。但需要注意的是,只有顺势疗法医生才能根据具体情况开出有效且安全的剂量。
預測
天使综合征的预后取决于染色体异常的性质及其检测的及时性。受影响最严重的是那些15号染色体基因缺失(缺失)的儿童。这类患者能够行走和说话的可能性极低。其他情况可以通过谨慎的治疗和对孩子的爱来纠正。
不幸的是,这些患者虽然不愚笨,能够理解语言及其含义,但无法成为社会的正式成员。然而,他们的沟通能力终生都会受到影响。患者可以从小学习手语,但不能强迫他们用语言交流。“会说话”患者的词汇量仅限于日常生活中使用的最低限度词汇(5-15个词)。
至于天使综合征患者的预期寿命和总体健康状况,此处的数据在平均值附近波动。成年期患者主要面临脊柱侧弯和肥胖等健康问题,但只要采取正确的治疗方法,这些问题都不会危及生命。