
亞急性壞死性腦肌病Leia
最近審查:23.04.2024

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
 [
[原因 leia綜合徵
該疾病的核心是缺乏能夠提供能量形成的酶,主要是由於丙酮酸代謝的破壞以及呼吸鏈中電子運輸的缺陷。丙酮酸脫氫酶複合物缺乏症的發展(A-E1亞基),丙酮酸羧化酶,絡合物1(NAD輔酶Q-還原酶)和絡合物4(細胞色素氧化酶)的呼吸鏈。
據發現缺陷丙酮酸鹽,絡合物1(NAD輔酶Q-還原酶)和絡合物4(細胞色素氧化酶)的呼吸鏈中以常染色體隱性方式遺傳,丙酮酸脫氫酶複合物(A-E1亞單位)的缺陷 - X連鎖隱性。在mtDNA點突變影響6-ATP酶亞基,線粒體遺傳特徵。最常發生與置換胸腺嘧啶與鳥嘌呤或胞嘧啶在位置8993相關的線粒體DNA突變mistsens。在線粒體DNA的9176位置罕見突變。由於這一事實,即突變T8993G - 在校正子NARP,在家庭用這兩種疾病的存在所描述的基本缺陷。孩子們還描述了線粒體DNA突變在位置8344,這是在綜合徵MERRF找到。
這表明,在大多數線粒體中突變mtDNA積累的情況下,Leia綜合徵的嚴重病程發展。在這種狀態的線粒體發生中,在所有線粒體的90%中檢測到突變mtDNA。發病機制與細胞內能量產生和乳酸酸中毒的發生有關。
症狀 leia綜合徵
這種疾病的第一個跡像是在幼年(1-3歲)初次登場。不過,也有6-7歲的在2個星期的疾病表現和案例。最初開發的非特異性疾病:精神運動發育遲緩,食慾不振,嘔吐發作,體重過輕。在隨後的生長神經症狀:肌張力減退或張力障礙與過渡到高滲,癲癇發作,肌陣攣性抽搐或強直 - 陣攣性發作,四肢,舞蹈手足徐動,協調障礙的震顫,腱反射減退,嗜睡,昏睡。腦神經變性具有進行性。錐體和錐體束外功能不全的症狀增加,吞嚥作用被破壞。常常有這樣的如眼瞼下垂,眼肌麻痺,視神經萎縮,視網膜色素變性少權威的變化。有時發生肥厚型心肌病,出現呼吸急促發作。
很少,疾病根據急性腦病的類型進行。更具特徵的是慢性或亞急性電流,在疾病發作幾年後導致致命的結果。隨著急流(幾週),由於呼吸中樞癱瘓而發生死亡。
診斷 leia綜合徵
在生物化學血液測試中,由於血液和液體中乳酸和丙酮酸的積聚以及血液中丙氨酸含量的增加,檢測到乳酸酸中毒。而且,酮體的水平可以增加。在尿液中,有機酸的排泄增加:乳酸,富馬酸等。血液和組織中肉鹼的水平通常降低。
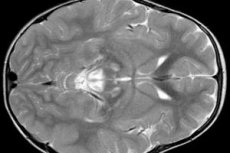
腦電圖結果顯示局灶性癲癇活動徵象。根據MRI數據,檢測到腦室擴張,雙側腦損傷,基底節(尾狀核,殼,黑色物質,蒼白球)的鈣化。還可以確定大腦半球和大腦物質的萎縮。
形態學研究顯示毛重變化腦實質:對稱壞死,脫髓鞘和腦海綿狀變性,主要是中間部位,橋樑,基底節,丘腦,視神經。組織學圖片包括腦組織囊變,星形細胞膠質增生,神經元死亡,細胞內線粒體數量增加。在骨骼肌 - 脂質的夾雜物的堆積減少到1,線粒體subsarkolemmalnoe擁塞的4呼吸鏈,異常線粒體嵴的破壞複合組織化學反應。RRF的現象往往未被發現。
如何檢查?
Использованная литература