
Charcot-Marie-Tooth 病。
最近審查:12.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
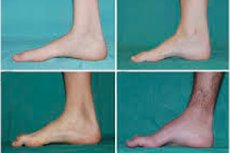
腓骨肌萎缩症、Charcot-Marie-Tooth 综合征或疾病是一组损害周围神经的慢性遗传性疾病。
根据ICD-10,该疾病在神经系统疾病部分的代码为G60.0(遗传性运动和感觉神经病变)。该疾病也被列入孤儿病名单。
流行病學
据临床统计,每10万人口中各类腓骨肌萎缩症的患病率为19例(另有资料显示,每2.5-10千人口中患病率为1例)。
CMT 1型约占病例的三分之二(每5,000至7,000人口中有一例),其中近70%与PMP22基因重复有关。全球有超过120万人患有此类疾病。
4 型 CMT 的发病率估计为每 10,000 名儿童中 1-5 例。[ 1 ]
原因 夏科-玛丽-牙病
根据多发性神经病综合征的分类,腓骨(腓骨)肌萎缩症、Charcot-Marie-Tooth 神经性肌萎缩症或 Charcot-Marie-Tooth 病(缩写为 CMT)是指基因决定的运动感觉性多发性神经病。[ 2 ]
也就是说,其发病原因是基因突变。根据基因变异的性质,该综合征可分为两大类:脱髓鞘型和轴突型。第一类包括腓骨肌萎缩症1型(CMT1),其病因是17号染色体上编码跨膜外周髓鞘蛋白22的PMP22基因发生重复。结果导致轴突鞘(神经细胞突起)节段性脱髓鞘,神经信号传导速度降低。此外,其他一些基因也可能存在突变。
轴突型为腓骨肌萎缩症2型(CMT2),该病影响轴突本身,并与位于1p36.22位点的MFN2基因的病理改变有关。该基因编码膜蛋白线粒体融合蛋白-2,该蛋白对于线粒体融合以及周围神经细胞内功能性线粒体网络的形成至关重要。CMT2有十几种亚型(具体基因突变)。
值得注意的是,目前已鉴定出一百多个基因,这些基因的损伤会通过遗传传递,导致各种类型的腓骨肌萎缩症。例如,RAB7基因突变会导致2B型腓骨肌萎缩症;SH3TC2基因(编码施万细胞的一种膜蛋白)突变会导致4C型腓骨肌萎缩症,这种疾病在儿童时期发病,其特征是运动和感觉神经元的脱髓鞘(该病4型有十几种类型)。
一种罕见的 3 型 CMT(称为 Dejerine-Sottas 综合征)在儿童早期开始发展,是由 PMP22、MPZ、EGR2 和其他基因的突变引起的。
当 CMT 5 型发生在 5-12 岁之间时,不仅会出现运动神经病(表现为下肢痉挛性截瘫),还会损害视神经和听觉神经。
肌肉无力和视神经萎缩(伴有视力丧失)以及平衡问题是 CMT 6 型的典型症状。而 7 型夏科-马里-图斯病不仅存在运动感觉性神经病变,还存在视网膜色素变性形式的视网膜疾病。
X 连锁 CMT 或称腓骨肌萎缩症,在男性中更为常见,伴有四肢轻瘫(双臂和双腿运动无力),是一种脱髓鞘疾病,被认为是由 X 染色体长臂上的 GJB1 基因突变引起的。该基因编码连接蛋白 32,这是施万细胞和少突胶质细胞的跨膜蛋白,负责调节神经信号的传递。[ 3 ]
風險因素
CMT 的主要危险因素是家族病史,即近亲有此病史。
据遗传学家称,如果父母双方均为腓骨肌萎缩症(Charcot-Marie-Tooth 病)的常染色体隐性基因携带者,其子女罹患此病的风险为 25%。而子女携带此基因(但不出现任何症状)的风险估计为 50%。
在X连锁遗传(突变基因位于女性的X染色体上)的情况下,母亲将该基因遗传给儿子的风险为50%,而儿子最终会患上CMT。女儿出生时可能不会发病,但女儿的儿子(孙辈)可能会继承该缺陷基因,并患上CMT。
發病
在任何类型的夏科-马里-图斯病中,其发病机制都是由周围神经的遗传异常引起的:运动神经(运动)和感觉神经(敏感)。
如果CMT类型是脱髓鞘性的,那么保护周围神经轴突的髓鞘的破坏或缺陷会导致周围神经系统(大脑、肌肉和感觉器官之间)的神经冲动传递减慢。
在轴突类型的疾病中,轴突本身受到影响,这会对神经信号的强度产生负面影响,不足以充分刺激肌肉和感觉器官。
另请阅读:
夏科-马里-图斯综合征是如何传播的?缺陷基因可以以常染色体显性或常染色体隐性的方式遗传。
最常见的类型是常染色体显性遗传,即突变基因只有一个拷贝(由父母一方携带)。据估计,每个出生的孩子遗传 CMT 的概率为 50%。[ 4 ]
对于常染色体隐性遗传,疾病需要两个缺陷基因的副本(来自每个未表现出任何疾病迹象的父母)才能发展。
40%-50% 的病例为常染色体显性遗传性脱髓鞘病变,即 CMT 1 型;12%-26% 的病例为轴突性 CMT,即 2 型。10%-15% 的病例为 X 连锁遗传。[ 5 ]
症狀 夏科-玛丽-牙病
通常情况下,这种疾病的最初症状在儿童和青少年时期开始出现,并在整个生命过程中逐渐发展,尽管这种综合症也可能在之后才显现出来。症状的组合多种多样,疾病的进展速度和严重程度都无法预测。
初期的典型症状包括全身疲劳加剧;足部、踝部和小腿肌肉张力下降(无力);反射消失。这会导致足部活动困难,并导致步态障碍(步态不稳),表现为双腿抬高,并经常绊倒和跌倒。幼儿患上腓骨肌萎缩症的症状可能包括明显的笨拙和与年龄不符的行走困难,并伴有双侧足下垂。足部畸形也是其特征:足弓高(凹足)或严重扁平足,以及弯曲(锤状)脚趾。
如果孩子在肌肉张力减退的背景下用脚趾行走,神经科医生可能会怀疑孩子患有 4 型 CMT,这种疾病的孩子可能在青春期无法行走。
随着病情进展,肌肉萎缩和无力会蔓延至上肢,导致精细运动技能和正常手部动作难以完成。触觉、冷热感知能力下降以及手脚麻木等症状表明感觉神经轴突受损。
在儿童时期出现的 3 型和 6 型 Charcot-Marie-Tooth 病中,可以观察到感觉性共济失调(运动和平衡协调受损)、肌肉抽搐和震颤、面神经损伤、伴有眼球震颤的视神经萎缩以及听力损失。
在后期,可能会出现无法控制的颤抖(震颤)和频繁的肌肉痉挛;运动问题可能导致疼痛的发生:肌肉、关节、神经性疼痛。
並發症和後果
腓骨肌萎缩症可引起以下并发症和后果:
- 更频繁的扭伤和骨折;
- 与关节周围肌肉和肌腱缩短有关的挛缩;
- 脊柱侧弯(脊柱弯曲);
- 呼吸问题——当支配横膈膜肌肉的神经纤维受损时:
- 丧失独立行动的能力。
鑑別診斷
鉴别诊断包括其他周围神经病、杜兴氏肌营养不良症、脊髓型肌无力综合征、糖尿病性神经病变、多发性脊髓侧索硬化症和肌萎缩性脊髓侧索硬化症、格林-巴利综合征、腓神经创伤及其萎缩(包括被夹在腰椎间盘之间时)、小脑或丘脑损伤,以及化疗的副作用(在使用长春新碱或紫杉醇等细胞抑制剂治疗期间)。[ 7 ]
誰聯繫?
治療 夏科-玛丽-牙病
目前,这种遗传性疾病的治疗包括运动疗法(旨在增强和拉伸肌肉);职业疗法(帮助手部肌肉无力的患者);以及使用矫形器械辅助行走。必要时,医生会开具止痛药或抗惊厥药。[ 8 ]
对于严重的扁平足,可以进行截骨术;对于足跟变形,则需要手术矫正——关节融合术。[ 9 ]
目前,针对该疾病的遗传因素及其治疗方法的研究正在进行中。使用干细胞、某些激素、卵磷脂或抗坏血酸等疗法尚未取得积极成果。
但得益于最近的研究,在不久的将来,腓骨肌萎缩症的治疗或许确实会出现一些新进展。因此,法国公司Pharnext自2014年以来一直在开发用于治疗成人1型腓骨肌萎缩症(CMT)的药物PXT3003,并于2019年中期开始进行临床试验。该药物可以抑制PMP22基因表达的增加,改善周围神经髓鞘形成,并缓解神经肌肉症状。
美国萨雷普塔治疗公司(Sarepta Therapeutics)的专家正在研发一种针对1型腓骨肌萎缩症(Charcot-Marie-Tooth disease)的基因疗法。该疗法将使用一种无害的Dependovirus属腺相关病毒(AAV),该病毒具有线性单链DNA基因组,可将NTF3基因导入体内,该基因编码神经营养因子3(NT-3)蛋白,而神经营养因子3是雪旺氏神经细胞正常功能所必需的。
Helixmith 将于 2020 年底开始对韩国开发的基因疗法 Engensis (VM202) 进行临床试验,以治疗 1 型 CMT 的肌肉症状。[ 10 ]
預防
预防CMT可以通过对未来父母进行遗传咨询来实现,尤其是在夫妻中有此病家族史的情况下。然而,也已发现新生点基因突变的病例,即家族史中没有此病的病例。
在怀孕期间,可以通过绒毛活检(妊娠 10 至 13 周)以及羊水分析(妊娠 15-18 周)检查未来孩子患有 Charcot-Marie-Tooth 病的可能性。
預測
总体而言,不同类型的腓骨肌萎缩症的预后取决于临床严重程度,但所有病例的病情进展均较为缓慢。许多患者会出现残疾,但这并不会影响预期寿命。