
颈椎和胸椎脊髓空洞症
最近審查:04.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
“脊髓空洞症”一词源自希腊语,字面意思是“脊髓空洞”。该病症是一种慢性中枢神经系统疾病,其特征是脊髓内形成充满液体的空腔。少数情况下,该疾病也会影响延髓。
脊髓空洞症是由神经胶质组织损伤或颅椎交界处畸形引起的。该疾病是众多无法治愈的疾病之一,可通过磁共振成像进行诊断。[ 1 ]
流行病學
脊髓空洞症是一种慢性进行性神经系统病变。该疾病的特征是脊髓(通常位于下颈段或上胸段)形成特殊的空洞,导致相应区域某些类型的感觉丧失。该问题也可能扩散至延髓。如果脑桥受累,则患者被诊断为延髓空洞症。腰部病变以及脊柱完全性病变极为罕见。
脊髓空洞症主要影响男性(约2:1)。临床症状通常在较年轻时(约25岁)发现,35-40岁时较少见。
超过一半的病例与 Arnold-Chiari 综合征有关。[ 2 ],[ 3 ]
真正的脊髓空洞症通常伴有脊柱的先天性畸形和发育缺陷,例如脊柱弯曲、胸部形态异常、咬合不正、颅面部和其他骨骼节段不对称、耳部发育不良、舌分叉、多指或乳头增多等。超过30%的病例为真正的脊髓空洞症,具有家族性,且多见于男性。其他所有脊髓空洞症病例均伴有颅椎连接处缺陷,导致椎管增宽。在椎管扩张最严重的区域,灰质会被破坏,从而引发特征性症状。其他病因包括脊髓损伤、出血和脊髓梗塞。
脊髓空洞症的患病率约为每十万人3例。一些研究表明,考虑到种族和地域差异,脊髓空洞症的患病率为每十万人8.4例至每十万人0.9例。[ 4 ],[ 5 ] 约75%的病例在青年和中年时期(20-45岁)出现劳动能力受限或丧失。[ 6 ]
原因 鞘膜积液
脊髓空洞症可以是先天性的,也可以是后天性的:
- 这种先天性畸形是由于胚胎时期脊柱和脊髓发育异常造成的。负责保护神经结构的神经胶质细胞成熟过慢,其中一些在中枢神经系统形成后仍继续生长。
- 获得性脊柱侧弯是肿瘤过程、痉挛、脊柱外伤以及急性感染性和炎症性疾病的结果。
上述任何一种脑病都伴有过多神经胶质细胞的形成。在这些神经胶质细胞不断死亡的背景下,形成了内衬神经胶质组织的空腔。液体很容易穿透这层屏障,因此空腔很快就会被脑脊液填满:形成囊性结构,并逐渐增大。下一步是附近结构的压力增加,导致疼痛综合征,四肢和身体各种感觉丧失。[ 7 ]
造成这种病理的主要原因被认为如下:
- 中枢神经系统先天性异常;
- 肿瘤扩散至脊髓结构和大脑下部;
- 脊柱创伤和发育异常;
- 椎管病理性狭窄;
- 颅底到脊柱的过渡区受损;
- 过度体力消耗。
如今,专家们仍在继续研究导致脊髓空洞症的风险因素。
風險因素
在导致脊髓空洞症发展的因素中,最重要的因素是:
- 呼吸系统的急性和慢性感染和炎症病理;
- 繁重的体力劳动;
- 影响脊柱的损伤、炎症和肿瘤过程;[ 8 ]
- 职业和生活条件不令人满意。
其他因素包括:
- 吸烟会显著增加脊柱出现问题的风险,因为它会导致血液中的氧气含量降低,从而导致组织出现营养障碍。
- 超重会给脊柱带来额外的压力。有时,只有减掉多余的体重才能缓解这种疾病的症状。
- 太高(男性超过180厘米,女性超过175厘米)。
發病
脊髓空洞症是由于脊髓结构发生紊乱而引起的。患者的脊髓内会形成微小的囊性区域。这些囊性区域周围会生长结缔组织(而非神经组织),从而压迫和破坏传递痛觉和温度觉的感觉通道。[ 9 ]
即使是先天性脊髓疾病,其病理变化的出现和进一步发展也主要由外部刺激引起。外部影响会导致内部疾病的出现,最终导致脊髓空洞症的发生。[ 10 ]
科学家们发现,大多数患有脊髓空洞症的人都是经常从事重体力劳动的人。生产机械化劳动的引入导致脊髓空洞症发病率下降,这一事实也证实了这一点。[ 11 ]
目前,以下因素越来越被认为是脊髓空洞症患者的病因:
- 过去的创伤,背部挫伤;
- 体温过低,长时间暴露在寒冷环境中;
- 吸烟、酗酒;
- 不关注自身健康,忽视疾病的最初迹象,自行用药,不及时就医。
在医学上,疾病的发病机制可分为以下几种:
- 由于胚胎发育阶段的失败而导致后颅窝和脊髓区域脑脊液循环中断;
- 髓管闭合不正确,形成后缝,这是由于骨缺损和神经胶质瘤病导致的,随后发生腐烂和囊性和裂隙性改变的形成。
遗传性体质性疾病表现为特定的脊髓闭合障碍特征,这些特征以常染色体显性遗传方式遗传,并代表着对病变的易感性。髓管和颅椎连接处发育缺陷为病变的发展提供了有利条件。[ 12 ]
脊柱和脊髓损伤以及身体微创伤都可能造成致病。脊髓损伤最常发生在颈椎和上胸椎区域,其次是下胸椎和腰骶椎区域。[ 13 ]
在一些患者中,病理过程扩散至延髓(以延髓空洞症的形式),较少见的是扩散至脑桥和内囊。[ 14 ]
症狀 鞘膜积液
大多数脊髓空洞症患者的脊髓后角区域都会形成空洞。负责痛觉和温度觉的敏感神经细胞就位于此处。在患者的皮肤上,可以发现一些敏感度尚不明确的区域。这些区域通常出现在手臂和身体上,类似于“半截夹克”和“夹克”,分别对应单侧和双侧损伤。
在此处阅读有关脊髓空洞症的症状和类型的更多信息。
並發症和後果
脊髓空洞症的并发症可能包括:
- 肌肉萎缩、挛缩;
- 继发感染,发展为肺炎、支气管肺炎、膀胱炎、肾盂肾炎;
- 感染进入伤口并损伤皮肤,发生化脓性过程,直至出现脓毒症并发症;
- 延髓麻痹的发展,可能导致患者呼吸衰竭和死亡。
专家指出,脊髓空洞症通常进展缓慢,很少发展为严重疾病。一种例外是该疾病的侵袭性进行性形式,其中脊髓空洞的形成持续进行。这种病症不仅对患者健康构成威胁,而且对患者的生命也构成威胁:需要紧急手术治疗。
脊髓空洞症的病程通常难以预测:该病的发病过程呈稳定期和进展期交替出现。病情进展可持续数周至数年,有时病情急剧恶化,有时又急剧减缓。在诱发因素(剧烈咳嗽、剧烈头部运动等)的影响下,先前无症状的患者也可能出现急性临床表现。
患者的生活质量与患有心力衰竭或恶性肿瘤的人相当。
可能的术后并发症包括:
- 脑脊液漏(水漏);
- 假性脑膜膨出;
- 分流位移;
- 暂时性神经功能缺损。
手术后此类并发症的发生率相对较低。
脊髓空洞症的主要后果是脊髓病,可发展为截瘫和四肢瘫痪,引起痉挛、褥疮和溃疡的形成、肺炎复发,还可导致肠道和泌尿生殖系统功能紊乱。[ 15 ]
診斷 鞘膜积液
诊断措施始于询问患者。医生应注意脊髓损伤和颅椎病变等症状群的特征性体征。可疑体征包括:
- 感觉障碍(感觉异常,疼痛,痛觉缺失,感觉减退,温度感觉下降);
- 手臂、颈部、后脑勺、胸部疼痛;
- 某些部位感到寒冷或寒冷,麻木;
- 持续性头痛、耳神经和视觉障碍(眼痛、畏光、复视、视力丧失、头晕、前庭疾病、耳内压力和噪音、听力损失、眩晕)。
在检查过程中,有必要向患者明确遗传因素、既往病史和损伤以及体力活动情况。由于脊髓空洞症的急性发作非常罕见,且该病通常进展缓慢且迁延不愈,因此有必要确定该病的大致发病时间。
在检查患者时,需要注意是否存在脊髓空洞症的典型临床表现:轻瘫、敏感性障碍、植物营养变化。
实验室测试不具有特异性,是作为一般临床研究的一部分规定的:
- 一般血液和尿液分析;
- 生化血液检查。
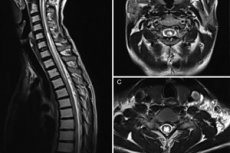
仪器诊断主要以MRI为代表。该技术使我们能够评估囊性结构的参数,描述其大小和形态。最佳方法是使用T1模式的矢状面投影,因为它对液体运动的敏感性较低。脊髓空洞症的典型MRI征象如下:
- 脊髓信号改变,如纵向、中央或旁中央区域,其强度与脑脊液的强度相似;
- 脊髓横向体积可能增加;
- 病理腔隙最常见的位置是颈胸区;
- 腔的分布范围从2个节段至脊髓的整个长度;
- 腔体直径 – 2-23 毫米;
- 当腔隙大小超过8毫米时,可观察到脊髓肿大。
建议沿脊柱的整个长度进行 MRI 检查。
腔体形状有以下几种:
- 对称,位于中心,圆椭圆形;
- 形状不规则,位于脊髓中央或近中央区域。
第二种类型的腔位于脊髓前动脉和脊髓后动脉之间的区域,与蛛网膜下腔没有连接,最常与外部损伤有关 - 例如创伤。
磁共振成像不仅在诊断阶段进行,而且还在治疗效果的动态监测中进行:
- MRI 显示空洞形成不完全(即所谓的“脊髓空洞前症”):脊髓扩张但无肿瘤,并伴有间质水肿;
- 空腔塌陷的MRI征象:空腔垂直方向变扁平,水平方向拉伸,伴有脊髓萎缩。
根据指征进行重复磁共振成像检查。如果病理相对稳定,可以每2年进行一次重复检查。
- 根据病变部位及其临床特征,对颅骨、颅椎区、脊柱、上肢和关节进行X光检查。脊髓空洞症可以识别骨骼发育缺陷、神经营养不良、骨质疏松灶、关节病、骨骼异常等。病理改变的程度有助于评估疾病的严重程度和预后。
- 计算机断层扫描 (CT) 的信息量不如核磁共振成像 (MRI) 或 X 光片。只有结合脊髓造影术和水溶性造影剂才能检测到病理性空洞。[ 16 ]
- 肌电图有助于明确脊髓前角运动神经元是否存在损伤,甚至有助于识别前角过程的临床前期问题。
- 通过神经肌电图,我们可以看到最初的锥体紊乱和轴突变性。
- 脑电图检查对于确定脑干结构的功能受损和延髓空洞症的最初迹象是必要的。
- 脑回声描记术用于检测脑空洞症并有助于识别脑内扩大的脑室系统。
- 感觉测量诊断用于明确敏感性障碍的位置和强度。
鑑別診斷
鉴别诊断根据以下病理和情况进行:
- 根据磁共振成像的结果可以确定髓内肿瘤(特别是影响颈椎时)和延髓肿瘤。
- 脊髓出血——其特征是损伤后立即出现急性症状,随后病情逐渐消退。脊髓空洞腔出血会使诊断变得复杂。
- 肌萎缩侧索硬化症的特点是发展速度快,并且在脊髓可视化过程中可以观察到病理变化的特征。
- 缺血性脊髓型颈椎病具有特定的发病病因,以动态原理上的敏感性障碍为特征,并有脊椎造影和MRI的特征性体征。
- 脊髓囊肿、肿瘤、创伤后或囊性脊髓病、脊髓蛛网膜炎、结核性脊柱炎。
- 颅椎缺损(寰椎及枢椎发育不全、扁平足、颅底压痕等)伴有神经系统症状,但未形成囊性结构。主要的鉴别诊断方法是MRI检查。
- 雷诺氏病、血管营养病。
- 压迫性缺血性神经病(腕管综合征或肘管综合征)。[ 17 ]
Arnold Chiari 畸形和脊髓空洞症需要鉴别诊断吗?这两种疾病通常并存:脊髓空洞的形成与小脑扁桃体移位有关,有时甚至与躯干和第四脑室移位至枕骨大孔水平以下有关。这些疾病的“元凶”通常是遗传因素,只有通过手术干预才能治疗。[ 18 ]
脊髓空洞症和脊髓积水需要进行强制性鉴别。脊髓鞘膜积水的特征是脑脊液量显著增加,且压力升高。脊髓积水常与脊髓空洞症并存,但需要注意的是,这两个术语并不相同,指的是两种不同的病理。诊断需根据临床、放射学和断层扫描数据进行。[ 19 ]
脊髓空洞症和延髓空洞症并非同义词。延髓空洞症是指脊髓空洞症的病理过程延伸至脑干区域,并伴有特征性症状:眼球震颤、延髓功能障碍以及面部局部分离性麻木。
誰聯繫?
治療 鞘膜积液
然而,脊髓空洞症最有效的治疗方法被认为是手术干预。当神经功能不全进展,尤其是出现下肢中枢性瘫痪或上肢周围性瘫痪时,必须进行手术。手术干预包括解剖中央椎管并进行引流。该手术确实有效:大多数患者术后病理发展停止,神经系统疾病减少。创伤后和感染后线性脊髓空洞症可通过在囊性结构和蛛网膜下腔之间进行分流来纠正。如果病理的根本原因是髓内肿瘤,则应切除肿瘤。小脑疝是颅后窝减压术的指征。
預防
预防脊髓空洞症的主要措施是避免可能扰乱脑脊液动力学的活动。重要的是尽量减少腹压和颅内压升高的可能性:不要提举重物,避免过度体力活动(包括强烈的静态压力)、剧烈咳嗽和打喷嚏、用力等。您还应避免脊柱和头部损伤,保持健康、适度活跃的生活方式。不宜缺乏运动。
如果已经确诊脊髓空洞症,则应采取预防措施,防止病情恶化。以下措施是强制性的:
- 药房神经科登记;
- 系统的诊断程序来监测病理动态(磁共振成像 - 每2年一次或更频繁,取决于适应症);
- 定期接受神经科医生的检查(每年 1-2 次)。
脊髓空洞症是一种动态病理,持续的临床观察和诊断措施有助于及时发现病理过程的恶化并采取适当的治疗措施。这一点至关重要,尤其是在儿童时期患有脊髓空洞症的情况下,正确评估手术治疗的适应症至关重要:如果脊髓空洞症的发展与骨骼系统的快速生长相关,则有些病例会自行痊愈。
預測
脊髓空洞症会损伤脊髓结构,导致身体和四肢的运动能力和感觉受损。痛觉和温度觉丧失可能导致严重损伤和烧伤。运动功能障碍伴有肌肉无力和萎缩。
反过来,脊髓空洞症会导致脊柱变形并恶化:患者常常会发展为脊柱侧弯。这种情况很少见,但也存在一些病变没有任何症状,而是在MRI检查中意外发现的情况。
脊髓空洞症的预后取决于临床表现的严重程度和范围、疾病持续时间及其病因。对于大多数患者来说,唯一有效的治疗方法是手术,以稳定脑脊液循环。手术干预的类型由神经外科医生决定。
大约每两个接受有效治疗的患者中,就有一个只出现轻微的病理改变。虽然有关于自发康复的信息,但这种结果仅在个别病例中发现,主要发生在儿科。这类病例是由骨骼的快速生长和脑结构自然空间扩张引起的。脊髓空洞症更常导致残疾。
大多数患者在长期病程的背景下会发展为不可逆的脊髓疾病,这使得术后预后恶化:许多症状即使在手术后仍然存在。然而,这并不意味着手术毫无意义且不合适:得益于这种治疗,有可能阻止疾病的进一步发展。