
阿珀综合征
最近審查:04.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。

 [
[ 發病
Apert综合征是一种遗传性疾病,属于常染色体显性遗传(即,如果一个家庭中父母一方患病,则其孩子罹患此病的概率为50%-100%)。
成纤维细胞生长因子受体2 (FGFR2) 的独特突变导致沿成骨途径发育的祖细胞数量增加。这最终导致胎儿发育过程中骨膜下骨基质形成增加,以及颅缝过早骨化。颅缝融化的顺序和速度决定了畸形和残疾的程度。一旦颅缝材料愈合,垂直于该颅缝的其他组织的生长就会受到限制,融合的骨骼将充当单一的骨支架。
第一个遗传证据是,Apert 综合征中的并指畸形是由于角质形成细胞生长因子受体 (KGFR) 缺陷引起的,即观察到成纤维细胞中 KGFR 的表达与并指畸形的严重程度之间存在相关性。
FGFR2 Ser252Trp 突变患者更易出现弱视和斜视,FGFR2 Pro253Arg 突变患者更易出现视盘萎缩。FGR2 Ser252Trp 突变患者的视力障碍患病率显著高于 FGFR2 Pro253Arg 突变患者。
症狀 阿珀综合征
由于该疾病在母亲子宫内发育,其部分症状在婴儿出生时即可明显显现。该综合征的主要症状包括:
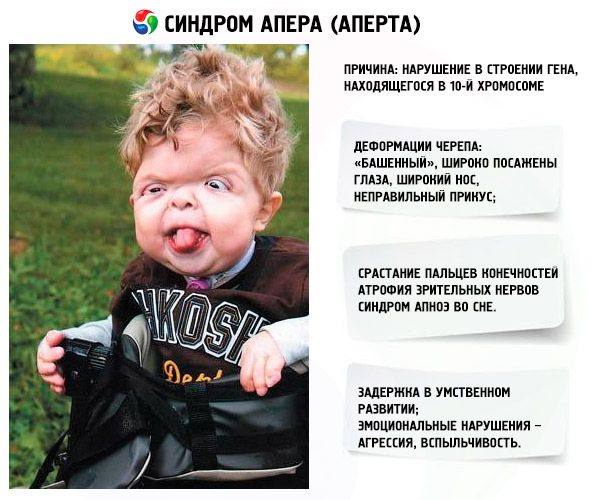
- 头骨变形 - 高度增加,呈现“塔”状;此外,眼睛距离较宽且略微凸出(因为眼窝缩小);鼻子变宽,咬合不正确(上牙过度突出);
- 四肢手指完全融合(主要是无名指,还有中指、食指),形似皮膜或骨骼完全融合;另外,可能长出多余的手指;
- 智力障碍(并非每个人都会出现);
- 视神经萎缩,导致视力下降(在某些情况下,甚至完全丧失视力);
- 由于颅缝过早闭合导致颅内压升高,表现为头痛和恶心呕吐;
- 由于上颌发育不全,出现呼吸问题;
- 睡眠呼吸暂停综合症很常见。
- 情绪表现——攻击性、缺乏克制、易怒。
診斷 阿珀综合征
为了做出诊断,需要采取以下步骤:
- 医生应分析患者的主诉以及病史。有必要了解家族中是否有过类似的病症;
- 神经系统检查以评估颅骨形状和患者的智力发育(专门问卷以及访谈);
- 检查眼底以发现是否存在颅内压升高的症状(视神经乳头肿胀以及边缘模糊);
- 为了评估颅骨的状况,需要进行 X 光检查;
- 通过头部计算机和磁共振成像逐层检查大脑和颅骨的结构,确定是否存在颅缝过早融合的症状,此外还有脑积水(由于颅内压升高,积聚了过多的脑脊液(这是促进代谢过程的脑脊液,以及脑的营养));
- 对脚和手进行 X 光检查以确定手指融合的原因(这对于计划后续手术干预很重要);
- 可能需要咨询医学遗传学家和神经外科医生。
测试
正在对FGFR2型基因中常见的突变进行基因分析。
鑑別診斷
该综合征必须与其他伴有颅缝早闭的遗传性疾病相鉴别。这些疾病包括Pfeiffer综合征、Crouzon综合征、Saethre-Chotzen综合征和Carpenter综合征。分子遗传学检测可用于排除这些异常。
誰聯繫?
治療 阿珀综合征
手术被认为是治疗 Apert 综合征的唯一有效方法——它有助于纠正个人的身体缺陷,还可以纠正智力障碍。
在此过程中,冠状缝会被闭合,以防止可能造成的脑损伤。最常见的技术是颅面牵引术,即分级颅骨牵引。正畸和/或正颌手术可用于去除个体面部缺陷。
此外,患者还需接受手指融合手术矫正。
参考
医学遗传学。国家指南。由 GEOTAR-Media 副总裁 Puzyrev Ginter EK 编辑。 2022年