
先天性動脈瘤
最近審查:07.06.2024

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
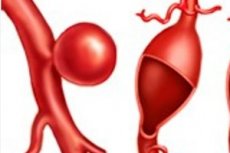
病理性弱化和随后由于先天性缺陷或遗传疾病而发生的动脉血管,心室或间隔的局部膨胀被诊断为先天性动脉瘤。
流行病學
综合动脉瘤占病例的10-15%;胸腔主动脉瘤和解剖发生在80%的Marfan综合征患者中。
根据研究,先天性心脏病中肺动脉动脉瘤的患病率约为病例的6%,成人先天性左心室动脉瘤的患病率不超过0.7%。心房间隔动脉瘤有1%的儿童和1-2%的成人。
主动脉鼻窦动脉瘤的发生率估计占普通人群的0.09%,占所有先天性心脏缺陷的3.5%。
大约50%的先天性脑静脉畸形患者中,大约50%的脑动脉瘤出现,但在12岁以下的儿童中,他们的病例不到5%。根据临床统计,所有颅内动脉瘤中有85%发生在大脑中威利斯圆的血管中。原因 先天性動脈瘤
注意到先天性动脉瘤的原因,专家首先将结缔组织的遗传性病理学放在:
中:- 马凡综合症;
- Ehlers-Danlos综合征 (血管类型);
- Loes-Dietz综合征。
动脉动脉瘤在患者中更容易形成 威廉姆斯综合征 (Williams-Boyren),这是由ELN基因的缺失引起的,该基因编码了Tropoelastin蛋白,Tropoelastin蛋白是弹性蛋白的前体(血管壁组织的细胞外基质的重要结构元素)。
)。非伴有遗传(家族性的)胸动脉瘤也被定义,某些基因(MYH11,ACTA2等)中的突变已经被鉴定出来。
先天性起源的动脉瘤可以在存在遗传酶异常的情况下形成 - 不同的类型 儿童的粘二糖 这是由代谢糖胺聚糖所需的酶的基因突变引起的(粘多糖)。
出生时存在的脑动脉瘤不仅与综合征胶原蛋白有关。在:中观察到了大脑动脉圆的大脑血管壁的这种局灶性凸起:- 动静脉畸形 - 一种先天性血管病理学,其本质违反了动脉和静脉血管的建筑学,导致正常血液流动变化;
- 常染色体显性多囊性肾脏疾病。
腹主动脉或肾动脉的先天性动脉瘤形成 神经纤维瘤病 1型,一种遗传疾病,是由于近一半的患者自发突变引起的。
静脉动脉瘤的原因(在临床实践中很少)被认为是宫内发育过程中形成的血管畸形(例如,在Servel-Martorell综合征中)以及在Klippel-Trenaunay综合征中静脉膨胀类型的静脉膨胀类型的静脉血管成年,已发现
另请阅读:
發病
Marfan综合征中主要血管动脉瘤的主要发病机制是FBN1基因中的突变,并且会损害血管壁纤维蛋白1的细胞外基质蛋白的产生。这改变了培养基的结构和机械性能,动脉血管的中鞘(Tunica培养基)。除了主动脉上升段的动脉瘤外,该综合征患者还具有主动脉瓣不足。
由于某些基因突变导致了胶原蛋白型I和III型 - 血管壁组织的胶原蛋白I和III纤维蛋白的脆弱性来解释
由于Ehlers-Danlos综合征的血管类型引起的动脉瘤形成的机制(包括脾动脉)。这种变化还削弱了血管壁,在最弱的位置或“血液动力胁迫”增加的血液流动性血流动力的作用下,它逐渐扩大并凸起。
与转化生长因子受体(TGF-β)基因突变相关的Loes-dietz综合征中动脉动脉瘤的发病机理,专家相信弹性蛋白的碎片和丧失,其纤维的纤维纤维的纤维纤维提供了大型血管壁的可逆性弹性。这些纤维的损伤导致容器壁的收缩性降低。同时,蛋白聚糖分子积聚在内侧细胞间基质中,从而降低了I型胶原蛋白的稳定性,从而维持血管壁的强度。
在粘多糖化中,在容器壁的内皮和内侧细胞中,有大量的糖胺聚糖,大的线性多糖(碳水化合物)破坏胶原纤维的排列。
除了进行性肾囊肿形成外,多囊肾脏疾病中的PKD1/PKD2突变被认为会导致胶原蛋白缺陷,并且这种遗传疾病的患者具有主动脉和囊肿头部的动脉解剖和动脉瘤 - 冠状动脉,冠状动脉,冠状动脉层间speclialialialialial和intracrialial and Intracrachanial and intracraclanial and。
近年来的组织学研究表明,在神经纤维瘤病1型的情况下,腹主动脉或肾动脉的动脉瘤形成可能是由外周神经系统的schwann细胞的增殖而引起的,这些神经系统的schwann细胞扩散到血管壁的鞘内。
在脑动脉血管缺陷中,脑动脉瘤形成的机制与血液流动分布的干扰有关:颈动脉(即,颈内动脉中的血液流动 - 颈内动脉中的血液流动)和基底力 - 基底 - 在脑部的主要动脉中,
症狀 先天性動脈瘤
根据它们的形状,有类型的动脉瘤,例如囊状和纺锤形(梭形),根据它们的定位,它们被分为类型:大脑(大脑血管动脉瘤),胸痛或腹部主动脉的动脉瘤, 外周动脉动脉瘤 和其他。
先天性主动脉瘤
动脉瘤影响主动脉瓣主动脉瓣到iliac动脉的任何部分。 [5] ]他们的症状在出版物中详细介绍:
先天性主动脉鼻动脉瘤
Valsalva的主动脉窦或窦是升主动脉的解剖学扩张之一 主动脉瓣 。主动脉窦动脉瘤位于主动脉瓣环和升主动脉区域之间,其中血管的构型再次变为管状(所谓的senotubular连接)。这种动脉瘤是由于血管壁的弹性层片而形成的,并形成为盲憩室。一个不充气的主动脉窦动脉瘤通常是无症状的,但是如果足够大,它可能会表现出心律不齐, 主动脉反流 ,心房颤动并导致 急性冠状动脉综合征 。 [6] ], [7] ]
先天性脑血管动脉瘤
小动脉瘤可能不会引起任何症状,但首先迹象是突然发作严重的头痛,恶心和呕吐。大动脉瘤可能会压在大脑结构上,这表现为瞳孔扩张和双视,眼睑下垂,一只眼睛上方和后面的疼痛,脸部的一侧麻木以及癫痫发作。 [8] ], [9] ]
材料中的更多信息 - 大脑血管的动脉动脉瘤
先天性心动脉瘤
出版物中的所有细节 - 急性和慢性心动脉瘤:心室,隔膜,染色后,先天性
先天性心房隔膜动脉瘤
心房隔室动脉瘤是心房隔膜的先天性畸形,心房隔膜是一种局部的囊样畸形,通常在椭圆形窝中出现,并且在心脏周期期间通常会凸起成一个或两个心房。 [10] ]
病理学可能与先天性心脏缺陷有关,尤其是开放的椭圆形窗户,瓣膜脱垂和开放性动脉桥,但在三分之一的情况下,它表现为一种孤立的异常。
主要症状中有:呼吸急促,尤其是在身体劳累的情况下;疲劳;下肢和腹部肿胀;心律失常和pal症;心脏喃喃自语(听时)。 [11] ]
先天性静脉动脉瘤
这些是罕见的畸形 - 大多数大静脉中可能发生的血管异常;最常见的定位是下肢(超过75%的病例),上肢(最多10%)。内颈静脉的动脉瘤(通常通常是主轴形)占病例的13%,三分之二的患者是儿童和青少年。 [12] ]
浅表静脉动脉瘤表现为柔软的皮下质量(通常是无痛),随着推动,咳嗽或哭泣而扩大。 [13] ]
並發症和後果
动脉瘤壁是损失正常生物力学特性的病理上改变的血管壁,并且其膨胀部位的可扩展性受到限制,因此任何动脉瘤的主要后果和并发症是破裂的。
出版物中的所有细节 - 胸腔和腹主动脉瘤破裂
在这种情况下,先天性脑血管动脉瘤的破裂伴随 亚蛛网膜下腔出血 ,导致小儿死亡率超过10%。
主动脉鼻窦动脉瘤的破裂是一种危险的并发症:右侧和非冠状窦的破裂通常会导致主动脉与右心房或右心流出区之间的联系,从而导致左至公路放电。这可能会导致右心室过载和右心力衰竭。心脏的先天性动脉瘤,尤其是左心室或心房隔膜的并发症,可能是外部血栓形成(由于血液停滞而引起的);凝块可能会破裂,并具有外围动脉栓塞和隐性中风的潜力。当这样的动脉瘤破裂时 心脏填塞 。
診斷 先天性動脈瘤
要诊断先天性动脉瘤,需要进行全面检查,其基础是工具诊断,其中包括:ECG和超声心动图;胸部的超声和计算机断层扫描;腹腔内器官动脉的超声;冠状动脉造影;和主动脉造影;对比心室;多尖的CT血管造影和MR血管造影; CT扫描颅底;经颅多相扫描等。
鑑別診斷
鉴别诊断可能具有挑战性。例如,心室隔膜动脉瘤应与大的间隔缺陷,心室肥大,法洛氏四元和Eisenmenger综合征区分开。和静脉动脉瘤应与静脉曲张和截短静脉畸形,血管瘤,喉咙和肠囊肿区分开。
誰聯繫?
治療 先天性動脈瘤
治疗通常始于有助于降低血压或放松血管以防止动脉瘤破裂的药物。还使用抗凝剂 - 防止血凝块形成。
阅读:
在有症状的先天性动脉瘤中,手术治疗是根据动脉瘤的定位和大小使用各种技术(包括血管内)进行的。在文章中阅读更多信息 - 动脉动脉瘤的手术
預防
作为当今可用的预防措施,专家指出:医疗和遗传咨询, 怀孕的遗传分析 , 也 先天性疾病的产前诊断 。
預測
不幸的是,对于所有先天性动脉瘤病例,都不能有良好的预后。例如,患有结缔组织病理遗传确定综合症的患者有多长时间?通常,Marfan综合征的平均存活率约为70年,Ehlers-Danlos综合征约50年,而Loes-Dietz综合征的平均存活率仅为35年以上。在大多数情况下,死亡是由于动脉瘤,脑出血或胸部/腹主动脉夹层破裂而发生的。
主动脉鼻动脉瘤破裂的患者通常在诊断后的一年内死亡 - 由于临床医生的充血性心力衰竭。