
尤瑟尔综合征
最近審查:04.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
Usher综合征是一种遗传性疾病,其症状为出生时完全失聪,并随着年龄增长逐渐失明。视力丧失与视网膜色素变性有关,这是一种视网膜色素变性的过程。许多Usher综合征患者还伴有严重的平衡问题。
原因 尤瑟尔综合征
Usher综合征I、II和III型为常染色体隐性遗传,而IV型被认为是X染色体异常。该综合征导致失明和失聪的原因尚未得到充分研究。据推测,该病患者对可能破坏DNA结构的成分高度敏感。此外,该病可能与免疫系统疾病有关,但目前尚不清楚该病的具体病因。
1989年,人们首次在II型糖尿病患者中发现了染色体异常,这或许能在未来帮助我们分离出致病基因。此外,我们或许还能在携带者中识别这些基因,并开发出专门的产前基因检测方法。
 [ 8 ]
[ 8 ]
症狀 尤瑟尔综合征
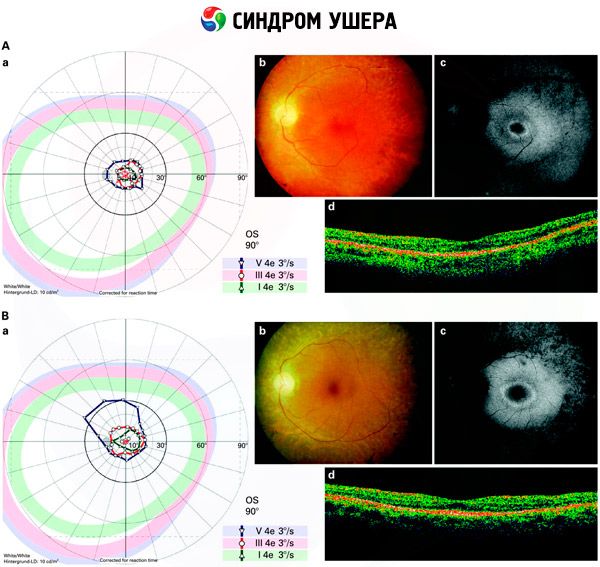
Usher综合征的症状包括听力丧失和眼部色素细胞异常积聚。随后,患者会出现视网膜退化,导致视力下降,在最严重的情况下最终丧失视力。
感音神经性听力损失可为轻度或完全性,通常不会在出生后发展。然而,视网膜色素病变可能在儿童期或之后开始出现。检查结果表明,即使周边视力恶化(这种情况称为“管状视野”),中心视力仍可维持多年。
这些是该疾病的主要表现,有时会伴有其他疾病,例如精神病和其他精神障碍、内耳问题和/或白内障。
形式
研究过程中,发现了该病的 3 种类型,以及一种较为罕见的第 4 种类型。
该疾病的I型特征是先天性完全性耳聋,并伴有平衡障碍。这类儿童通常在1.5岁时才开始行走。视力通常从10岁开始下降,最终发展为夜盲症,这种情况在20岁时开始出现。患有此类疾病的儿童可能会出现周边视力逐渐下降的情况。
II 型疾病表现为中度或先天性耳聋。在这种情况下,部分耳聋通常不再恶化。色素性视网膜炎通常在青春期末期或 20 岁后开始出现。夜盲症通常在 29-31 岁时出现。II 型病变患者的视力受损通常比 I 型病变进展得稍慢。
该病的 III 型特征是进行性听力损失,通常始于青春期,并在同一时期(稍晚于听力损失)逐渐发展为视网膜色素变性,这可能成为进行性失明的一个因素。
IV型病理表现主要见于男性。此类患者还会出现进行性病变以及听力和视力丧失。这种类型非常罕见,通常与X染色体有关。
診斷 尤瑟尔综合征
Usher 综合征的诊断是根据患者突发性耳聋和进行性视力丧失的症状组合做出的。
测试
可能需要进行特殊的基因测试来检测突变。
已发现 11 个基因位点可导致 Usher 综合征的发展,并且已确定 9 个基因肯定是导致该疾病的病因:
- 类型 1:MY07A、USH1C、Cdh23、Pcdh15、SANS。
- 类型 2:ush2a、VLGR1、WHRN。
- Usher 综合征 3 型:USH3A。
NIDCD 的科学家与来自纽约和以色列大学的同事一起,发现了 Pcdh15 基因中一种名为 R245X 的突变,这种突变导致了犹太人群中很大一部分 1 型 Usher 综合征患者患病。
要了解进行临床试验的实验室,请访问 https://www.genetests.org 并在实验室目录中搜索“Usher 综合征”。
要了解包括 Usher 综合征基因检测在内的现有临床试验,请访问 https://www.clinicaltrials.gov 并搜索“Usher 综合征”或“Usher 综合征基因检测”。
[ 25 ]、[ 26 ]、[ 27 ]、[ 28 ]、[ 29 ]、[ 30 ]
仪器诊断
仪器诊断的方法有多种:
- 检查眼底以检测视网膜上是否存在色素斑以及视网膜血管是否变窄;
- 视网膜电图,可以检测视网膜的早期退行性偏差。它显示了放射电通路的消失;
- 眼震电图 (ENG) 可测量可能表明存在不平衡的不自主眼球运动。
- 听力测验,用于确定是否存在耳聋及其严重程度。
鑑別診斷
Usher 综合征必须与一些类似的疾病相区别。
霍尔格伦综合征,其特征是先天性听力损失和进行性视力丧失(也可能出现白内障和眼球震颤)。其他症状包括共济失调、精神运动障碍、精神病和智力障碍。
阿尔斯特罗姆综合征是一种遗传性疾病,患者视网膜退化,导致中央视力丧失。该综合征与儿童肥胖有关。同时,患者10年后会开始出现糖尿病和听力损失。
孕妇在妊娠初期感染风疹可能会导致胎儿出现各种发育异常。这种异常的后果包括听力损失、视力问题(或视力问题),以及各种发育缺陷。
誰聯繫?
治療 尤瑟尔综合征
目前,Usher 综合征尚无治愈方法。因此,该病的治疗主要包括减缓视力丧失的进程,以及弥补听力损失。可能的治疗方法包括:
- 服用维生素 A(一些眼科医生认为,高剂量的维生素 A 棕榈酸酯可能会减缓但不能阻止视网膜色素变性的进展);
- 将特殊的电子设备植入患者的耳朵(助听器、人工耳蜗。
眼科医生建议,大多数患有常见类型视网膜色素变性的成年人在医生指导下每日服用15,000 IU(国际单位)维生素A棕榈酸酯。由于1型Usher综合征患者未纳入研究,因此不建议这类患者服用高剂量维生素A。正在考虑服用维生素A的患者应与医生讨论此治疗方案。其他关于此治疗方案的建议包括:
- 改变饮食习惯,摄入富含维生素 A 的食物。
- 计划怀孕的女性应在计划怀孕前三个月停止服用高剂量的维生素 A,因为这会增加出生缺陷的风险。
- 由于出生缺陷的风险增加,怀孕的妇女应停止服用高剂量的维生素 A。
帮助这样的孩子适应社会生活也很重要。这需要特殊教育教师和心理学家的帮助。如果患者的视力开始逐渐丧失,应该教他使用手语。
預測
Usher综合征的预后不佳。大多数患有此病的患者,无论何种类型,其视野和视力都会在20-30岁时开始下降。在某些情况下,甚至会出现双侧完全失明。听力损失通常伴有听力障碍,并很快发展为双侧完全失聪。