
遺傳和代謝方面的骨關節炎的發病機制
最近審查:23.04.2024

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
機械因素在骨關節炎發病機制中的作用是不容置疑的,但有令人信服的數據表明某些形式的骨關節炎是根據孟德爾法則遺傳的。遺傳性骨關節病可分為:
- 原發性廣泛性骨關節病(PGOA),
- 晶體相關的關節病,
- 由於遺傳性骨軟骨發育不良造成的早期骨關節炎。
1803年,W.Heberden在刷子的遠側指間關節的背面描述了“略微緻密的結,小豌豆的大小”。據該作者稱,這一特徵將骨關節炎與包括痛風在內的其他關節疾病區分開來。J. Hayagarth(1805)擴展了Geberden節點的臨床描述,注意到他們頻繁與其他本地化的關節病相關。Bouchard進一步描述了手部近側指間關節背側表面上的類似節點。使用術語“Heberden和Bushard節點”,W. Osier分享了“肥大性關節炎”和“變形性關節炎”(1909)。1953年,RM Stecher和H. Hersh發現Heberden節點在家庭成員中的分佈,並得出結論,他們以常染色體顯性方式遺傳。在發現RM Stecher和H. Hersh之後,研究揭示了Geberden和Bushard節點與其他關節退行性病變的關聯。根據目前的臨床檢查和HLA分型,JS Lawrence(1977),JS Lawrence和合著者(1983)提出存在多基因遺傳,而不是單個基因的缺陷。
遺傳性骨關節炎的表型譜在臨床上僅在達到成年後期才表現出輕微形式,在兒童期表現出非常嚴重的形式。傳統上,所有這些形式都被歸類為繼發性骨關節炎。它是目前已知,對於某些這些表型的是編碼破壞軟骨基質的完整性和軟骨細胞增殖和基因表達的調節關節軟骨ECM大分子的基因的突變。這些遺傳性疾病代表了骨關節炎的一個亞群,它不同於繼發性骨關節炎。
遺傳性和繼發性骨關節炎之間的差異(根據Williams CJ和Jimenez SA,1999)
|
遺傳性骨關節炎 |
繼發性骨關節炎 |
|
|
病因 |
在關節軟骨中表達的基因突變 |
各種遺傳性和獲得性疾病 |
|
發病 |
損傷關節軟骨的結構或功能組分 |
疾病的次要表現,並不總是只影響關節軟骨 |
|
治療 |
可能的基因治療用於糾正基因缺陷 |
治療潛在的疾病 |
軟骨發育不良/骨軟骨發育不良 - 一組臨床異質性疾病,特徵在於關節軟骨和生長板的生長和發育異常。一些HD / OXD導致骨關節炎的早期發展,臨床特徵為嚴重的過程。其中,可以區分以下疾病:
- 脊椎動脈發育不良(SED),
- 綜合徵Stickler,
- 克尼斯納發育不良,
- 多發性epiphysical發育不良(MED),
- 形而上學軟骨發育不良(MHD),
- 一些oto-spondylo-metaepiphysial dysplasias(OSMED)。
遺傳性不典型增生的特點是早期發生骨關節炎(根據Williams CJ和Jimenez SA,1999)
|
病 |
軌跡 |
繼承類型 |
突變的基因 |
突變類型 |
|
早髮型遲髮型SED(SAR)* |
12q13.1-q13.2 |
FROM |
COL 2 A, |
鹼基替換,插入,刪除 |
|
Stickler綜合症(STL1) |
12q13.1-q13.2 |
FROM |
COL2A1 |
更換底座,插入 |
|
Stickler綜合症(STL2) |
6r21.3 |
FROM |
COLA |
插入,刪除 |
|
Stickler綜合症 |
1r21 |
FROM |
COLA |
更換底座 |
|
瓦格納綜合症 |
12q13.1-q13.2 |
FROM |
COUA, |
更換底座 |
|
OSMED |
6r21.3 |
美聯社 |
COLA |
更換底座 |
|
馬歇爾綜合症 |
1r21 |
FROM |
COLA |
插入 |
|
發育不良腹瀉 |
12q13.1-q13.2 |
FROM |
COLA |
插入,刪除 |
|
M3fl(EDM1) |
19r13.1 |
FROM |
COMP |
更換底座 |
|
BIP(EDM 2) |
1r32.2-RZZ |
FROM |
COLA |
插入 |
|
MHDShmida(MCDS) |
6q21-q22.3 |
FROM |
COLA |
基地的替代,刪除 |
|
MXD Yansena(MCDJ) |
Zr21.2 r21.3 |
FROM |
PTHR, |
更換底座 |
*括號內是軌蹟的符號; AD - 常染色體顯性; AR是常染色體隱性。
脊椎動脈發育不良
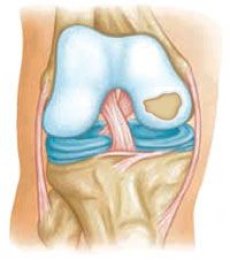
Spondiloepifizialnye發育不良(DMS)包括與常染色體顯性遺傳疾病,其特徵在於所述中軸骨骼的發育異常和長骨的骨骺的重變化,往往造成侏儒症一組異質性。通常,SED臨床上難以進行,伴隨著身體縮短和四肢減少。
SED的形式在較晚的時候表現出來,表型通常幾乎沒有變化,並且在嚴重骨關節炎發展之前可能不會臨床出現,直到青春期。腰椎變形可能表現為椎間盤狹窄,platipondylia和無意義的後凸畸形。此外,外周關節的骨骺異常及其早期退行性改變。外周關節病變的最常見徵像是踝關節和膝關節關節面的扁平化,以及股骨髁間溝的扁平化。通常存在股骨頭部和頸部的異常,伴隨著髖關節骨關節炎的發展,這表現在青春期。
由於這樣的事實,II型膠原蛋白-透明軟骨的ECM的主要成分已被建議,EDS的原因被編碼基因COL1A。早期骨關節炎的晚期艙單SED相關聯的表型,和膠原II型基因COL之間的遺傳聯繫的第一描述2 A,適用於1989年和1990年首次報導的突變COL 2 A,與後期清單相關聯的早期骨關節炎親屬SED ,關注Arg519> Cys鹼基的置換。迄今為止,已經鑑定了四個具有相似突變的家族。其他家庭早期OA易流動EDS檢測鹼基替換Arg75>半胱氨酸的成員,雖然SED-表型在這個家庭的成員不是在位置519與EDS家庭的代表類似家庭用替換精氨酸到半胱氨酸的表型也發現其他突變COL 2 A-Gly976> Ser,Gly493> Ser。J.施普蘭格爾等人(1994)使用的術語“類型11 kollagenopatii”來描述遺傳性疾病軟骨初級突變類型II膠原基因COL1A。
經典形式的Stickler綜合徵
它在1965年由GB Stickler及其合著者首次描述,他們將其稱為遺傳性關節病 - 眼病。綜合徵描述的GB綜合徵的特徵是視力器官損傷和嚴重的退化性關節病,這種疾病通常發生在生命的第三或第四個十年。這是一種常染色體顯性疾病,其發病率約為每1萬名新生兒中的1人。該疾病的臨床表現包括近視,進行性耳聾,腭裂,下頜發育不全(皮埃爾 - 羅賓的異常)和表皮發育不全。在新生儿期,在Stickler綜合徵患者的X光片上,骨骺增大,主要是股骨近端和脛骨遠端。在生長過程中,骨骺發育不良發展,表現為骨骺骨化不規則和隨後的退行性改變。
由於COL 2 A,在關節軟骨,和眼球的玻璃體表達,該基因斯蒂克勒綜合徵相關的外觀的病理學。然而,一些家庭提供斯蒂克勒綜合徵的調查顯示,並非所有的家庭疾病與COL關聯2 A.疾病稱為I型斯蒂克勒綜合徵(符號STL1基因)的這種形式。
Stickler綜合徵的臨床表現譜差異很大,目前已經確定了幾種表型。其中 - 瓦格納綜合徵,其特點是眼球失敗的盛行; 瓦格納綜合徵中的OA實際上並沒有發展,儘管COL 2 A 基因的突變(Gly67> Asp鹼基的替代)在患者中被揭示。目前尚不清楚為什麼這種突變COL只能影響玻璃體的功能,並不影響透明軟骨。
Stickler綜合徵的另一種形式是所謂的荷蘭變種; 除了對視覺器官的損害之外,其特徵在於該綜合徵的所有經典表現。HG Brunner等(1994)表明,與突變COL基因相關的荷蘭人斯蒂克勒綜合症的表型,A 2:顯性突變是隨後通過缺失外顯子的M.Sirko-Osadsa的54個鹼基對的缺失等人(1998)報導的另一個家庭描述無關的先前的作者,具有相似的表型和基因突變COL,以及2(缺失的27個鹼基對),其支持數據HG Brunner等(1994)。這種變體被稱為Stickler的II型綜合徵(基因座STL1的符號)。
最近,Stickler綜合徵的第三個基因座在患有玻璃體和視網膜病變的家族成員中發現,其與典型“綜合徵”中觀察到的變化在表型上顯著不同。這個家族的代表發現了COL2A基因的突變 (鹼基Gly97> Val的取代)。當然,為了證實AJ Richards和共同作者的發現,需要對Stickler綜合徵的這種表型和基因型病例進行新的描述。
很長一段時間討論了馬歇爾綜合徵和經典版的斯特勒氏綜合徵的統計學問題。現在馬歇爾綜合徵被歸類為單獨的表型,主要是由於面部骨骼更明顯的變形,儘管外周關節損傷與I型綜合徵相似。在馬歇爾綜合徵中,30年後開始出現膝關節和腰骶椎骨關節炎。綜合徵的原因是COL n A1 型膠原蛋白IX基因的突變。
OSMED
這種表型在荷蘭家庭進行了說明,其中在關節退行性變化,類似於骨關節炎出現在青春期和主要影響,髖,膝,肘和肩關節的成員; 還發現了鮮明的五官,增加腰椎前凸,增加指間關節,聽力喪失,但沒有透露視力任何異常器官(Vikkula M.等,1995)。研究人員已經發現的基因的編碼突變2 α-鏈II型膠原COL ,,甲2。
發育不良腹瀉
它的特徵是軀乾和四肢縮短,鼻子麵部和背部變扁,突眼和關節嚴重異常。在Knin綜合徵患者中,關節通常在出生時大,在童年和青春期早期會繼續增加。他們還經常可以檢測近視,聽力喪失,腭裂,馬蹄內翻足; 大多數患者出現早期嚴重的退行性改變,特別是在膝關節和髖關節表達。在脊柱的X線圖上,可以檢測出椎體扁平化和相當長的伸長,platiponylia。長的管狀骨變形為啞鈴,骨骺骨化減慢。在手的關節中,骨骺變平,關節變窄。關節軟骨軟,彈性降低; 組織學上發現大囊腫(“瑞士奶酪”的症狀)。Knyst綜合徵的原因是COb2A1型前膠原II基因的突變。
 [7], [8], [9], [10], [11], [12], [13], [14]
[7], [8], [9], [10], [11], [12], [13], [14]
多發性骺發育不良(MED)
一組異質的,其特徵在於長骨的生長板的發育異常,以及早期(體現在童年)嚴重的骨關節炎影響軸向和周邊縫(通常膝蓋,臀部,肩部和手的關節)的疾病。臨床DER為疼痛和僵硬的關節表現,步態的變化。患者DER還表現出在脊柱最小的變化(不同程度的椎體的扁平化的),有時骨架完好。它還有一個特點是身材矮小的患者,雖然侏儒症是罕見的。視覺器官不受影響。DER包括幾個選項,如費爾班克斯和羅紋表型。
MEDs以常染色體顯性類型遺傳,具有不同程度的外顯率。由於DER異常生長板的標誌,已經提出的是,這些基因的原因是有缺陷的發育不良編碼大分子軟骨生長板。結果發現至少有三個位點與DER表型有關。研究E.J. Weaver等人(1993),從“元兇”DER基因排除JT赫克特及其同事(1992)膠原II型和VI,蛋白聚醣和軟骨蛋白偶聯的核心蛋白。JT赫克特及其同事(1993),R. Oehelmann等人(1994)發現了DER和臨床上與之接近綜合徵psevdoahondroplazii和19號染色體著絲粒的區域之間的關聯。隨後的研究已經確定了3例編碼軟骨寡聚基質(OMPH)的蛋白EDR(符號EDM1座)的基因的突變。由於所有三個突變都發生在大概編碼鈣結合域OMPH基因的區域是這種蛋白質的鈣結合功能對於軟骨生長板的正常發展至關重要。
MD Briggs等(1994)報導了家庭荷蘭的,將其用1號染色體的部分相關聯的,包括類型IX膠原COL1A1(EDM軌跡符號的基因之一DER-表型2)。值得注意的是,所觀察到的突變型膠原IX的作用的第一個證據是局部的膠原原纖維II的表面上,在保持透明軟骨的完整性。迪爾M。等人(1995)表明,費爾班克斯的表型的基因不與任何軌跡EDM ,,沒有軌跡EDM相關聯2,這證實了DER的異質性。
形而上學軟骨發育不良(MHD)
異質的(描述超過150種)的一組透明軟骨的遺傳性疾病的,這是臨床上表現早期骨關節炎。MCH的特徵是乾骺端骨骼的改變。臨床上他們表現身材矮小,腿短,小腿彎曲,“鴨”步態。此外,在患者與其他系統(如免疫和消化)的參與MHD跡象顯現。軟骨生長板,其在組織學上明顯的簇增殖和肥大軟骨細胞的觀察到的解體,通過在軟骨下骨增厚隔片和紊亂的矩陣,和滲透nekaltsifitsirovannogo軟骨包圍。
Jansen,Schmid和McCusick的綜合徵是研究得最深的MHD。它們在骨骼異常特徵方面相似,但嚴重程度不同(Jansen綜合徵-McKusick綜合徵 - Schmid綜合徵)。最常見的是Schmid綜合徵(MCDS基因座的象徵),其由常染色體顯性類型遺傳。X線綜合徵表現為髖內翻,管狀骨縮短和彎曲,幹骺端杯狀變形(在近端比在遠端股骨更明顯)。在長管狀骨的生長板中觀察到最明顯的變化。
Schmid綜合徵患者至少有17種不同的X型膠原基因突變。膠原X型在生長板的肥大軟骨細胞中表達,並且可能參與骨化過程。因此,編碼膠原蛋白的X型COb2A1基因的突變是Schmid's綜合徵最可能的原因。
Jansen綜合徵患兒有高鈣血症,尿中磷酸鹽水平升高,甲狀旁腺激素(PG)和PG連接肽水平降低。由於後者的異常,可能是Jansen綜合徵的出現。1994年,AS Karaplis和合著者發表了原始研究的結果。編碼小鼠胚胎幹細胞中PG連接的肽的基因被破壞後,該等位基因缺陷的小鼠在出生後立即死亡。它們在軟骨下骨的發育中存在異常,這違反了軟骨的生長,並且軟骨細胞的增殖減少。1995年,E.Schipani和合著者報導了Jansen綜合徵患者PG連接的肽受體基因的雜合突變。該突變包括取代Gys223> Arg的鹼基,導致cAMP的積累; 這意味著223位氨基酸組氨酸在信號傳遞中起著至關重要的作用。後來,E.Schipani和合著者(1996)報導了另外三名Jansen綜合徵患者,其中兩名患者俱有相似的突變,第三位患者替代TruA10> Pro 。
原發性廣泛性骨關節炎
遺傳性原發性全身骨關節炎的最常見的形式是骨關節炎(非洲採礦夥伴關係),其最初被描述為單獨的疾病分類學JH Kellgren R.摩爾並在1952年的臨床原發性全身骨關節炎特性外觀布沙爾節點和赫伯登,多關節病變。原發性全身骨性關節炎的特點是骨關節炎和他的進展迅速體現的早發。射線照相原發性全身骨關節炎不從非遺傳性關節炎不同。儘管原發性全身骨性關節炎的發病機理的問題仍在討論,研究已經證實遺傳傾向的發生和原發性全身骨關節炎的發展具有重要作用。
所以,JH Kellgren等人(1963)發現Busharai赫伯登節點在男性親屬的36%,和女性親屬的49%,而數字分別17和在總人口中的26%。原發性骨關節炎的廣義越來越發現HLA單倍型A1V8和MZ-亞型A1抗胰蛋白酶。在雙胞胎TD斯佩克特和他的同事(1996)的一個經典研究中進行的膝關節的X射線和手的在130單和雙卵120雙女性的接頭的變化骨關節炎的特徵的存在。據發現骨關節炎的那一致影像學證據的所有站點用雙卵和遺傳因素促成介於40〜70%相比,在單卵雙胞胎高2倍。進行GD Wright等(1997)研究結節骨關節炎表現出疾病的嚴重程度和疾病的發病年齡之間呈高度負相關的早期發病的患者的年齡和受孕的父母。
在晶體相關性關節病中,關節腔內尿酸和含鈣晶體的晶體沉積具有家族傾向。
遺傳性晶體相關性關節病(根據Williams,C.J。和Jimenez SA,1999)
|
病 |
軌跡 |
繼承類型 |
突變的基因 |
突變類型 |
|
痛風(HPRT)* |
Xq27 |
與X染色體相關聯 |
HPRT1 |
基地的替代,刪除 |
|
痛風(PRPS) |
Xq22-Q24 |
與X染色體相關聯 |
PRPS1 |
更換底座 |
|
原發性焦磷酸鹽關節病(CCAL1) |
5r15.1-r15.2 |
FROM |
? |
? |
|
焦磷酸鹽關節病與早期發作的0A(CCAL2) |
8Q |
FROM |
? |
? |
*括號內是軌蹟的符號; AD是常染色體顯性。
1958年,D. Zintann S. Sitaj提出了27例病人的臨床病理描述,稱為“軟骨間鈣化”。大多數患者屬於5個家族,這表明該疾病的發病機制中存在遺傳成分。後來,D. McCarty和JL Hollander(1961)報導了兩名懷疑痛風伴隨關節腔內非永久晶體沉積的患者。X線檢查發現許多關節的透明軟骨異常鈣化。
射線照相焦病焦磷酸鈣關節病的二水合物結晶沉積或類似零星OA,但是,它常常會影響關節,不是典型地用於常規形式osteoartrozaa(例如,掌指,舟形梁,髕骨 - 股骨膝蓋司)。當焦磷酸關節病常形成軟骨下骨囊腫。雖然在大多數情況chondrocalcinosis二次骨關節病的表現形式之前發生,在一些個體中的疾病可以開始特發性關節炎,這伴隨著代謝紊亂(血色素沉著症,甲狀旁腺機能亢進,gipomagnezemiya等人)中。
很可能,關節軟骨ECM的結構變化誘導二水合焦磷酸鈣晶體的沉積。AO Bjelle(1972,1981)中,來自瑞典與膠原蛋白的含量焦磷酸鹽關節病降低和膠原纖維的斷裂關節軟骨基質家族的中間區域中。由於這些網站不包含晶體,作者建議,所描述的基質異常可能會使他們的沉積和關節退行性改變的發展。基於焦磷酸的關節病K. Ishikawa等人(1989),I. Masuda等人(1991)的散發病例研究的基礎上得出的結論是起因chondrocalcinosis是編碼ECM的蛋白的基因的突變。CJWilliams等人(1993),AJ雷吉納托等人(1994)發現了一個雜合突變COL 2 A,(取代的鹼Argl5>的Cys)患有嚴重的早期骨關節炎ankilozirovaniya,遲發性spondiloepifizialnoy發育不良的臨床表型的大家族的成員和chondrocalcinosis透明和纖維狀軟骨。然而,事實證明,這個家族的成員與OA有關的繼發性軟骨軟骨病。
還有人認為ECM的無機組分促進了晶體的形成。例如,低鎂血症通過抑制焦磷酸酶而引起軟骨珊瑚病的發展,這進而減少了晶體的溶解。在焦磷酸鹽關節病患者的滑液中,檢測到無機磷酸鹽含量增加。這和其他觀察結果表明,在焦磷酸鹽關節病患者中,焦磷酸鹽代謝存在局部干擾。描述了核苷三磷酸焦磷酸水解酶的酶,其可能參與在其在ECM中沉積的區域中焦磷酸鹽晶體的形成。在散發病例,焦磷酸鹽關節病中觀察到的酶的含量增加,但沒有觀察到疾病這樣的異常的家族型(賴安LM等人,1986)。然而,當患者的家族焦磷酸鹽關節病培養的成纖維細胞和淋巴母細胞檢測到的無機磷酸鹽,這也支持有關本地代謝紊亂焦磷酸鹽在疾病的發病機理中的作用的假說的水平升高。
近年來,嘗試了確定基因導致家族性病例的焦磷酸關節病,“有罪”。所以,遺傳物質從與焦磷酸鹽關節病(緬因州,美國)的大家族的成員中獲得的分析,其中chondrocalcinosis開發繼發於嚴重急進骨關節炎nedisplasticheskomu,排除與該疾病基因座COL連接2。然而,這項研究的作者發現所研究表型焦磷酸鹽關節病和軌跡之間的關聯,位於染色體8(軌跡符號SSAL)的長臂。AG Hughes等人(1995)發現主chondrocalcinosis的表型之間的關聯在來自英國和軌跡CCAL1 ,,其染色體5上的該區域5r15短臂局部家庭。通過CJ Williams等人(1996),基因座CCAL1在家族成員與焦磷酸鹽關節病多個本地化近端日提交來自阿根廷比前面的情況-在5r15.1區域。來自法國的家庭成員發現了類似的基因型。
因此,所述研究的數據表明焦磷酸鹽關節病的家族形式是由至少三種不同基因的突變引起的臨床和遺傳異質性疾病。