
目前对高尿酸血症发病机制的看法
最近審查:07.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
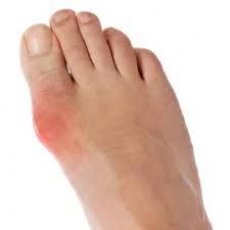
痛风是一种全身性痛风石性疾病,其特征是环境和/或遗传因素导致的高尿酸血症患者体内单尿酸钠晶体沉积在各个器官和组织中,并由此引起炎症。痛风的发病机制是基于尿酸(嘌呤)代谢紊乱和血液中尿酸(UA)含量的升高。尿酸代谢的基础是肾脏尿酸生成过多和排泄减少。同时,仅有10%的原发性痛风患者仅有内源性尿酸生成障碍。在其他患者中,高尿酸血症形成的主要因素是肾脏尿酸排泄障碍。
痛风除了损害肌肉骨骼系统外,还伴有内脏症状,其中之一就是尿酸盐肾病。尿酸盐肾病是慢性肾小管间质性肾炎的一种变体,其特征是尿酸晶体在间质中积聚,并伴有继发性炎症过程,同时肾小管上皮受损,导致其功能和重吸收过程受损。
肾脏运输尿酸的过程由四个级联过程组成:肾小球滤过、滤过尿酸几乎完全重吸收、分泌以及近端小管的排泄后重吸收。尿酸不与蛋白结合,因此可在肾小球中自由滤过。肾小管分泌尿酸的速度远低于肾小管重吸收的速度,因此分泌尿酸占总排泄尿酸量的比重很小。几乎98%-100%的滤过尿酸在近端小管重吸收,之后50%的滤过尿酸被重新分泌,然后几乎80%的排泄尿酸被重吸收,最终约7%-10%的滤过尿酸被排出。重吸收、分泌和分泌后重吸收阶段发生在近端小管。重吸收和分泌过程由位于近端小管上皮刷状缘的特定分子(转运蛋白)进行。
大多数尿酸转运蛋白属于OAT家族。尿酸的肾小管重吸收由一种名为URAT1(由SLC22A12基因编码)的有机阴离子转运蛋白(尿酸阴离子交换蛋白)完成。该转运蛋白仅存在于人类中。大量研究(包括针对家族性低尿酸血症患者的研究)表明,编码URAT1转运蛋白的SLC22A12基因存在突变。研究发现,丙磺舒和吡啶酰胺(一种具有抗尿酸排泄作用的抗结核药物)对这些患者的尿酸排泄几乎没有影响。
除了URAT1之外,还有其他转运蛋白:URATv1、SLC5A8编码的钠依赖性反向转运蛋白、OAT家族的有机阴离子转运蛋白(OAT1和OAT3、OAT2和OAT4)、ABCG2(集合管中的尿酸转运蛋白)、SLC2A3(近端小管的钠/磷酸盐反向转运蛋白)。OAT2和OAT4位于近端小管的顶端膜上;OAT1和OAT3位于其基底外侧部分,其主要功能是交换有机阴离子和双羧酸盐,但同时也有关于其对尿酸转运影响的数据。
URATv1(OATv1),后来被命名为GLUT9,由SLC2A9基因编码,是一种电压依赖性的有机离子转运体,主要转运葡萄糖和果糖,同时也是一种尿酸转运体,该基因的多态性与低尿酸血症有关,这已在遗传学研究中得到证实。
影响尿酸分泌的机制研究较少。尿酸分泌受损与ATP依赖性泵的变化以及编码尿调节蛋白(Tamm-Horsfall蛋白,ABSG2基因)形成的MRP4基因突变有关。尿调节蛋白影响尿酸分泌的确切机制尚不清楚,可能与近端小管钠重吸收增加以及尿酸同时增加有关。
肾脏转运蛋白受损,尿酸重吸收增加,可能导致高尿酸血症,并最终引发痛风。许多关于尿酸转运蛋白功能障碍的研究发现了基因突变,但这些研究大多集中于低尿酸血症患者尿酸转运蛋白是否存在基因突变,而高尿酸血症患者是否存在基因突变的问题仍未得到深入研究。值得注意的是,高嘌呤饮食、动脉高血压和局部缺血会激活URAT1和GLUT9转运蛋白,进而导致尿酸重吸收增加。有证据表明,通过URAT1进行的肾小管顶端尿酸和钠重吸收受损,在糖尿病酮症酸中毒、乙醇胺中毒、吡嗪酰胺治疗、高胰岛素血症和代谢综合征的影响下,随后会发展为高尿酸血症。因此,肾脏尿酸排泄受损可能是由于肾小管装置受损而导致的次要过程。
痛风患者肾小管功能可以通过每日尿酸排泄量、清除率、排泄分数 (EF)、尿酸重吸收量、钙 (Ca)、磷 (P) 和氨排泄量来评估。此外,患者的“标准”检查无法识别肾功能障碍的体征。最简单易行的方法是评估尿酸清除率,然后将其重新计算到体表面积。我们对痛风患者的研究表明,该测试在识别尿酸盐肾病体征方面具有相当高的信息含量,因此尿酸清除率低于 7 ml/min/1.73 m2 的灵敏度为 90%,特异性为 66%。
医院治疗系研究生Khalfina Tamila Nilovna。高尿酸血症发病机制的现代观点//实用医学。2012年12月8(64)/第1卷
 [
[