
多发性肌炎和皮肌炎:病因、症状、诊断和治疗
最近審查:05.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
 [
[ 什么原因导致皮肌炎和多发性肌炎?
该疾病的病因被认为是遗传易感人群对肌肉组织的自身免疫反应。该疾病在有家族病史和某些HLA抗原(DR3、DR52、DR56)携带者中更为常见。病毒性肌炎和恶性肿瘤是其可能的诱因。有报道称,在肌肉细胞中检测到了类似于小核糖核酸病毒的结构;此外,病毒也可在动物中诱发类似的疾病。恶性肿瘤与皮肌炎的关联性(远低于多发性肌炎)表明,肿瘤生长也可能是该疾病发展的诱因,因为它引发了对肿瘤和肌肉组织共同抗原的自身免疫反应。
IgM、IgG和补体第三种成分沉积于骨骼肌血管壁;这在儿童皮肌炎中尤为常见。多发性肌炎患者也可能出现其他自身免疫性疾病。
皮肌炎和多发性肌炎的病理生理学
病理变化包括细胞损伤和萎缩,并伴有不同程度的炎症。上、下肢以及面部肌肉的损伤程度较其他骨骼肌轻。咽喉和食道上段的内脏肌肉损伤,以及心脏、胃或肠道损伤(较少见)可导致上述器官功能障碍。横纹肌溶解症导致的肌红蛋白浓度升高可导致肾脏损害。关节和肺部也可能出现炎症变化,尤其是在存在抗合成酶抗体的患者中。
皮肌炎和多发性肌炎的症状
多发性肌炎的发病形式可为急性(尤其儿童)或亚急性(成人更常见)。急性病毒感染有时先于或诱发本病,最常见的症状是近端肌肉无力或皮疹。疼痛程度较无力轻。患者可能出现多关节痛、雷诺氏现象、吞咽困难、肺部疾病以及全身症状(体温升高、体重下降、虚弱)。雷诺氏现象常见于合并结缔组织疾病的患者。
肌肉无力症状可能持续数周或数月。然而,肌肉无力的临床表现至少需要50%的肌肉纤维受到影响(因此,肌肉无力的出现提示肌炎的进展)。患者可能难以将手臂抬高过肩、难以上楼梯或难以从坐姿起身。由于骨盆和肩胛带肌肉严重无力,患者可能只能依靠轮椅或卧床。如果颈部屈肌受到影响,则无法将头部从枕头上抬起。咽喉和食道上段肌肉受到影响会导致吞咽障碍和反流。下肢、上肢和面部肌肉通常不受影响。但可能会出现肢体挛缩。
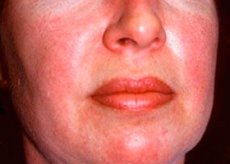
皮肌炎的皮疹通常颜色深且呈红斑状。眶周水肿呈紫色(向紫色皮疹)也是其特征性表现。皮疹可能略高于皮肤表面,表面光滑或有鳞屑;皮疹好发于额头、颈部、肩部、胸部、背部、前臂、小腿下部、眉毛、膝盖区域、内踝、指间关节和掌指关节背面以及外侧(Gottron症状)。指甲根部或周围可能出现充血。手指外侧皮肤可能出现脱屑性皮炎,并伴有皲裂。原发性皮损通常可消退且不留后遗症,但可能导致继发性病变,如色素沉着、萎缩、瘢痕形成或白癜风。可能会出现皮下钙化,尤其是在儿童中。
约30%患者出现多关节痛或多关节炎,常伴有水肿、关节积液,但关节表现程度较轻,多见于患者体内检测出Jo-1或其他合成酶抗体时。
多发性肌炎累及内脏器官(咽喉和食管上段除外)的情况较其他风湿性疾病(尤其是系统性红斑狼疮和系统性硬化症)少见。罕见情况下,尤其是在抗合成酶综合征中,该病表现为间质性肺炎(以呼吸困难和咳嗽为特征)。患者可能出现心律失常和传导障碍,但通常无症状。伴有血管炎的儿童更易出现胃肠道症状,包括呕血、黑便和肠穿孔。
哪裡受傷了?
多发性肌炎的分类
多发性肌炎有 5 种类型。
- 原发性特发性多发性肌炎可发生在任何年龄,且不涉及皮肤。
- 原发性特发性皮肌炎与原发性特发性多发性肌炎相似,但涉及皮肤。
- 多发性肌炎和皮肌炎伴发的恶性肿瘤可发生在任何年龄的患者中;其发展最常见于老年患者以及患有其他结缔组织疾病的患者。恶性肿瘤的发展可在肌炎发作前2年内和发作后2年内观察到。
- 儿童多发性肌炎或皮肌炎与系统性血管炎有关。
- 多发性肌炎和皮肌炎也可发生在患有其他结缔组织疾病的患者中,最常见的是进行性系统性硬化症、混合性结缔组织病和 SLE。
将躯干肌肌炎归入多发性肌炎是不正确的,因为后者是一种独立的疾病,其临床表现与慢性特发性多发性肌炎相似。然而,躯干肌肌炎多发于老年,常影响身体远端部位的肌肉(例如上肢和下肢),病程较长,治疗反应较差,并具有典型的组织学特征。
皮肌炎和多发性肌炎的诊断
对于有近端肌无力(无论伴或不伴压痛)症状的患者,应怀疑多发性肌炎。对于出现类似鸡血红蛋白征或Gottron征的皮疹的患者,以及有多发性肌炎表现并伴有任何与皮肌炎相符的皮肤病变的患者,均需评估皮肌炎的诊断。多发性肌炎和皮肌炎的临床表现可能类似于系统性硬化症,或不太常见的系统性红斑狼疮(SLE)或血管炎。尽可能满足以下五项标准可提高诊断的准确性:
- 近端肌肉无力;
- 特征性皮疹;
- 肌肉组织酶活性增加(肌酸激酶,或在其活性没有增加的情况下,氨基转移酶或醛缩酶);
- 肌动图或 MRI 的特征性变化;
- 肌肉组织活检中的特征性组织学变化(绝对标准)。
肌肉活检可以排除一些临床上相似的疾病,例如躯干肌炎和病毒感染引起的横纹肌溶解症。组织学检查发现的变化可能有所不同,但慢性炎症、肌肉退化和再生灶是典型特征。在开始潜在毒性治疗之前,必须进行准确的诊断(通常通过组织学验证)。MRI 可以检测肌肉中的水肿和炎症灶,然后进行针对性的活检。
实验室检查可以确诊或排除对该疾病存在的怀疑,还有助于评估其严重程度、与其他类似病症合并的可能性以及诊断并发症。尽管有些患者检测到了抗核抗体,但这种现象在其他结缔组织疾病中更为典型。约 60% 的患者有针对细胞核抗原 (PM-1) 或整个胸腺细胞和 Jo-1 的抗体。自身抗体在该疾病的发病机制中的作用仍不清楚,尽管已知抗 Jo-1 抗体是抗合成酶综合征(包括纤维化肺泡炎、肺纤维化、关节炎和雷诺现象)的特异性标志。
定期评估肌酸激酶活性有助于监测治疗效果。然而,对于严重肌萎缩的患者,即使存在慢性活动性肌炎,酶活性也可能正常。MRI、肌肉活检或肌酸激酶活性升高通常有助于区分多发性肌炎复发和糖皮质激素性肌病。
由于许多患者患有未确诊的恶性肿瘤,一些作者建议对所有60岁以上的皮肌炎和多发性肌炎成人患者进行筛查,筛查程序如下:体格检查,包括乳房、盆腔和直肠检查(包括粪便潜血检测);全血细胞计数;血液化学检查;乳房X光检查;癌胚抗原检测;尿液分析;胸部X光检查。一些作者质疑,对于没有恶性肿瘤临床证据的年轻患者,是否有必要进行此类筛查。
需要檢查什麼?
如何檢查?
皮肌炎和多发性肌炎的治疗
在炎症缓解前,应限制体力活动。糖皮质激素是一线治疗药物。在疾病急性期,成年患者应口服泼尼松龙,剂量为每日40至60毫克。定期测定肌酸激酶活性是疗效的早期指标:大多数患者在肌肉力量增加后6至12周内会观察到活性降低或恢复正常。酶活性恢复正常后,应减少泼尼松龙剂量:首先在一周内每天减少约2.5毫克,然后加快剂量;如果肌肉酶活性升高复发,则应再次增加激素剂量。康复患者可以不用糖皮质激素,但大多数成年患者需要长期糖皮质激素治疗(每日10-15毫克泼尼松龙)。儿童泼尼松龙的初始剂量为30-60毫克/平方米,每日一次。如果儿童病情缓解 > 1 年,可以停止糖皮质激素治疗。
在某些情况下,接受高剂量糖皮质激素的患者会突然出现肌肉无力症状,这可能与糖皮质激素性肌病的发展有关。
如果对糖皮质激素治疗反应不佳,或出现糖皮质激素性肌病或其他并发症,需要减少剂量或停用泼尼松龙,则应使用免疫抑制剂(甲氨蝶呤、环磷酰胺、硫唑嘌呤、环孢素)。部分患者可能仅接受甲氨蝶呤治疗(剂量通常超过类风湿性关节炎治疗剂量),治疗时间可能超过5年。静脉注射免疫球蛋白可能对药物治疗无效的患者有效,但会增加治疗费用。
原发性及转移性肿瘤相关的肌炎,以及躯干肌肌炎通常对糖皮质激素治疗较为难治。恶性肿瘤相关的肌炎在肿瘤切除后可能得到缓解。
皮肌炎和多发性肌炎的预后如何?
超过半数接受治疗的患者在五年内可获得长期缓解(甚至临床康复);儿童患者的比例更高。然而,复发可能随时发生。五年总生存率为75%,儿童患者更高。成人患者死亡的原因是严重且进行性的肌肉无力、吞咽困难、营养不良、吸入性肺炎或肺部感染导致的呼吸衰竭。如果多发性肌炎伴有心脏和肺部损伤,病情会更加严重,且难以治疗。儿童患者可能死于肠道血管炎。该病的总体预后也取决于是否存在恶性肿瘤。