新发现有助于更好地了解雷特综合征的病因
最近審查:02.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。

雷特综合征是一种罕见的神经发育障碍,目前尚无治愈方法或有效疗法。该病会导致严重的身体和认知症状,其中许多症状与自闭症谱系障碍相似。
雷特综合征是由 MECP2 基因突变引起的,该基因在大脑中高度表达,似乎在维持神经元健康方面发挥着重要作用。该基因位于 X 染色体上,雷特综合征主要影响女孩。为了开发雷特综合征的治疗方法,研究人员希望更好地了解 MECP2 及其在大脑中的功能。
包括怀特黑德研究所联合创始人鲁道夫·耶尼施(Rudolf Jaenisch)在内的研究人员几十年来一直在研究MECP2,但关于该基因的许多基本信息仍然未知。该基因编码的蛋白质MECP2参与基因调控;它与DNA结合,影响其他各种基因的表达水平,或它们产生的蛋白质量。
然而,研究人员并没有一份受 MECP2 影响的基因的完整列表,而且对于 MECP2 如何影响这些基因也没有达成共识。
MECP2 的早期研究表明它是一种抑制因子,可以降低其靶基因的表达。但 Jaenisch 等人此前的研究则表明,MECP2 也是一种激活因子,可以增加其靶基因的表达——而且它可能本身就是一种激活因子。此外,MECP2 的作用机制,或者说该蛋白质究竟是如何引起基因表达变化的,目前仍不清楚。
技术的限制阻碍了研究人员对这些问题的进一步探究。但 Yanish、其实验室的博士后 Yi Liu 以及 Yanish 的前实验室成员、现任蒙特利尔大学 CHU Sainte-Justine 研究中心助理教授的 Anthony Flamier,利用尖端技术解答了这些关于 MECP2 的遗留问题,并对其在大脑健康和疾病中的作用获得了新的见解。
他们的研究结果发表在《神经元》杂志上,研究人员还创建了 MECP2 数据的在线存储库,即MECP2-NeuroAtlas 门户,作为其他研究人员的资源。
“我认为这篇论文将从根本上改变人们对 MECP2 导致雷特综合征的理解。我们对这一机制有了全新的认识,这可能为开发这种疾病的治疗方法提供新的途径,”同时也是麻省理工学院生物学教授的 Janisch 说道。
深入了解大脑中的 MECP2
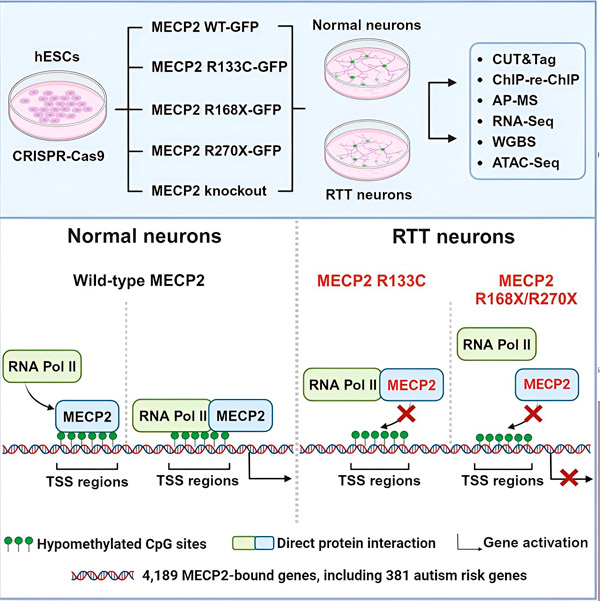
研究人员首先绘制了MECP2在人类神经元基因序列中结合位置的详细图谱,这些结合位置包括基因内部或基因附近DNA的调控区域。他们使用了一种名为CUT&Tag的方法,可以高精度地定位蛋白质与DNA的相互作用。
研究人员发现了超过4000个与MECP2相关的基因。他们在与雷特综合征相关的常见MECP2突变的神经元中重复了这些基因的映射,以确定在疾病状态下MECP2在哪里被耗尽。
了解了MECP2与哪些基因结合,刘和弗莱米尔开始将MECP2的靶点与大脑健康联系起来。他们发现,它的许多靶点都与神经元轴突和突触的发育和功能有关。
他们还将自己的 MECP2 靶点列表与西蒙斯基金会自闭症研究计划 (SFARI) 的自闭症相关基因数据库进行了比较,发现该数据库中有 381 个基因是 MECP2 靶点。
来源:Neuron (2024)。DOI:10.1016/j.neuron.2024.04.007
这些发现可能有助于阐明雷特综合征自闭症症状的潜在机制,并为研究 MECP2 在自闭症中可能发挥的作用提供一个良好的起点。
“我们绘制出了第一张MECP2表观基因组在健康和疾病状态下的整合图谱,这张图谱可以指导未来的研究,”刘教授说道。“了解哪些基因是MECP2的靶点,以及哪些基因在疾病中直接受到干扰,为理解雷特综合征以及探究神经元基因调控机制奠定了坚实的基础。”
研究人员还观察了MECP2是增加还是减少了其靶基因的表达。与MECP2被一些人认为是激活剂,另一些人则认为是抑制剂的历史相一致,刘和Flamier发现了MECP2同时扮演两种角色的例子。
然而,尽管MECP2通常被认为是一种抑制剂,但刘和弗莱米尔发现它主要是一种激活剂——这证实了Jaenisch和刘先前的研究结果。一项新的实验表明,MECP2至少能激活80%的靶蛋白,而另一项实验发现,它能激活高达88%的靶蛋白。
研究人员绘制的靶基因图谱进一步揭示了MECP2作为激活剂的作用。他们发现,对于MECP2激活的基因,它通常与基因上游的DNA区域(称为转录起始位点)结合。
细胞机制在此启动基因转录为RNA的过程,随后RNA被翻译成功能性蛋白质,而蛋白质是基因表达的产物。MECP2位于转录起始位点(基因表达的起点),这与它作为基因激活因子的作用相符。
研究人员随后着手确定 MECP2 在基因激活中的作用。他们观察了 MECP2 在这个位点除了 DNA 之外还能与哪些分子结合,发现 MECP2 直接与一种名为 RNA 聚合酶 II(RNA Pol II)的蛋白质复合物相互作用。RNA Pol II 是将 DNA 转录成 RNA 的关键细胞机器。RNA Pol II 无法自行找到基因,因此需要多种辅助因子(或称蛋白质协作者)来帮助它完成工作。
研究人员认为,MECP2 正是其中一种辅助因子,帮助 RNA 聚合酶 II 在 MECP2 结合的基因上启动转录。MECP2 的结构分析已鉴定出该分子与 RNA 聚合酶 II 结合的部分,其他实验也已证实,MECP2 的缺失会降低 RNA 聚合酶 II 在适当转录起始位点的存在,并降低靶基因的表达水平。
这表明,雷特综合征可能是由 MECP2 基因突变导致其靶基因转录减少引起的,而 MECP2 突变会导致其无法与 RNA 聚合酶 II 或 DNA 结合。与此观点一致,与疾病相关的最常见 MECP2 突变是截短突变:部分蛋白质缺失的突变,这可能会改变 MECP2 与 RNA 聚合酶 II 之间的相互作用。
研究人员希望,他们的发现不仅能改变我们对 MECP2 的理解,而且对 MECP2 如何影响大脑发育和功能的更深入和更广泛的理解可以带来新的见解,帮助患有雷特综合征和相关疾病(包括自闭症)的人。
“这个项目是 Janisch 实验室合作精神的绝佳体现,”Flamier 说道。“我和 Rudolf 遇到了一个与雷特综合征相关的具体问题,而我拥有 CUT&Tag 技术的经验,这项技术可以解决这个问题。通过讨论,我们意识到可以携手合作,现在我们拥有了一个关于 MECP2 及其与疾病关系的庞大信息库。”