发现防止骨质疏松症患者骨质流失的关键蛋白质
最近審查:02.07.2025

所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
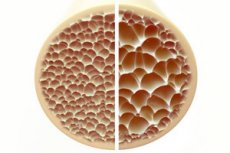
骨质疏松症是一种以骨骼疏松易碎为特征的疾病,对骨骼健康构成严重威胁。骨骼作为人体的主要结构支撑,提供至关重要的支撑。骨量减少不仅会削弱这种支撑作用,还会损害整体功能,导致生活质量下降。
随着老龄化人口中骨质疏松症发病率的上升,长期护理的医疗资源负担日益加重。因此,有必要了解骨质疏松症的发病机制,并开发有效的针对性治疗方法,以最大限度地减少其长期影响。
成骨细胞和破骨细胞是两种在骨组织维护和重塑中发挥关键作用的细胞。成骨细胞是负责新骨组织合成和沉积的骨形成细胞,而破骨细胞是参与分解和去除老化或受损骨组织的骨降解细胞。
破骨细胞比例增加会导致骨质流失,例如骨质疏松症、类风湿性关节炎(关节炎症)和骨转移(癌症扩散到骨骼)。破骨细胞源自巨噬细胞或单核细胞(属于免疫细胞)的分化。
因此,抑制破骨细胞分化可作为预防骨质流失的一种治疗策略。然而,调控骨重塑这一复杂过程的精确分子机制仍不清楚。
在一项新研究中,东京理科大学的Tadayoshi Hayata教授、Takuto Konno先生和Hitomi Murachi女士与其同事深入研究了破骨细胞分化的分子调控。核因子κB受体激活剂配体(RANKL)的刺激可诱导巨噬细胞分化为破骨细胞。
此外,骨形态发生蛋白 (BMP) 和转化生长因子 (TGF)-β 信号通路也参与调控 RANKL 介导的破骨细胞分化。在本研究中,研究人员旨在探究 Ctdnep1 的作用。Ctdnep1 是一种磷酸酶(一种去除磷酸基团的酶),据报道可以抑制 BMP 和 TGF-β 信号通路。
该研究发表在《生化与生物物理研究通讯》杂志上。
Hayata教授表示:“RANKL在破骨细胞分化中扮演着‘加速器’的角色。开车不仅需要油门,还需要刹车。在这里,我们发现Ctdnep1在破骨细胞分化中扮演着‘刹车’的角色。”
研究人员首先检测了RANKL处理的小鼠巨噬细胞和未处理的对照细胞中Ctdnep1的表达。他们观察到,Ctdnep1的表达在RANKL刺激下没有变化。然而,它在巨噬细胞中以颗粒形式定位于细胞质中,并分化为破骨细胞,这与它在其他细胞类型中正常的核周定位不同,表明它在破骨细胞分化中起着细胞质功能。
此外,Ctdnep1 的敲低(基因表达下调)导致抗酒石酸酸性磷酸酶 (TRAP) 阳性的破骨细胞数量增加,其中 TRAP 是分化破骨细胞的标志物。
Ctdnep1 敲除导致关键分化标志物表达增加,包括“Nfatc1”,这是一种由 RANKL 诱导的、促进破骨细胞分化的主要转录因子。这些结果支持 Ctdnep1 具有“制动功能”,即其负向调控破骨细胞分化。此外,Ctdnep1 敲除还导致磷酸钙吸收增加,表明 Ctdnep1 在骨吸收中起抑制作用。
最后,尽管Ctdnep1敲除并未改变BMP和TGF-β信号通路,但Ctdnep1缺陷细胞中磷酸化(活化)蛋白水平升高,而这些蛋白是RANKL信号通路的产物。这些结果表明,Ctdnep1对破骨细胞分化的抑制作用可能并非通过BMP和TGF-β信号通路介导,而是通过下调RANKL信号通路和Nfatc1蛋白水平介导。
总体而言,这些结果为破骨细胞分化过程提供了新的见解,并确定了潜在的治疗靶点,可用于开发减少破骨细胞过度活跃导致的骨质流失的治疗方法。除了以骨质流失为特征的疾病外,Ctdnep1还被确定为髓母细胞瘤(一种儿童脑肿瘤)的致病因素。作者们乐观地认为,他们的研究可以扩展到骨代谢以外的其他人类疾病。
Hayata教授总结道:“我们的研究结果表明,Ctdnep1是防止破骨细胞过度生成所必需的。这些结果可能进一步扩展我们对磷酸化-去磷酸化网络如何控制破骨细胞分化的认识,并可能为治疗与破骨细胞活性过度相关的骨病提供新的治疗策略。”